|
|
| (12 intermediate revisions not shown) |
| Line 5: |
Line 5: |
| | Related Links:</div> | | Related Links:</div> |
| | <div id="boxcontent"><DL> | | <div id="boxcontent"><DL> |
| - | <div class="heading">Vitamin A:</div>
| + | <div class="heading">Modeling:</div><DL> |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Project/VitA">Project</a><br/>
| + | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/Platforms">Modeling Platforms</a><br/> |
| - | <DD><a href="#">Parts</a><br/>
| + | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/LBSMod">LBS Models</a><br/> |
| - | <DD><a href="#">Protocols</a><br/></br/></DL>
| + | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/Opt">Optimization</a><br/> |
| - | <div class="heading">Modeling:</div><DL>
| + | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/Sensitivity">Sensitivity</a><br/> |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/Platforms">Modeling Platforms</a><br/>
| + | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/ParaFit">Parameter fitting</a><br/></DL> |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/Methods">Analytic Methods</a><br/>
| + | |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/GeneExp">Gene Expression</a><br/> | + | |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/VitA">Vitamin A</a><br/> | + | |
| - | <DD><a href="https://2011.igem.org/Team:Johns_Hopkins/Modeling/VitC">Vitamin C</a><br/></br/></DL> | + | |
| | </div> | | </div> |
| | </div> | | </div> |
| Line 20: |
Line 16: |
| | </html> | | </html> |
| | __NOTOC__ | | __NOTOC__ |
| - | ====== LBS: A New Approach to Modeling in iGEM ====== | + | ====== Introduction ====== |
| - | ===== Anatomy of an LBS model: the beta-carotene pathway (vitamin A) =====
| + | Once we sketched out schematic graphical models of vitamin A and vitamin C production, we set out to find the perfect modeling software. We wanted something simple, expressive, powerful, and extendable. We started where [https://2010.igem.org/Team:Edinburgh/Modelling/Kappa last year's modeling prize winner] left off: [http://kappalanguage.org/ Kappa]. |
| - | ==== Compartments and species ====
| + | |
| - | Here we declare two compartments under the root "world" compartment. First is the cytosol, inside of which goes the nucleus.
| + | |
| | | | |
| - | We specify ribosomes and RNA polymerases globally because they are shared across all gene expression processes. We then declare the crtE gene which codes for the enzyme GGPP synthase, the crtYB gene which codes for the bifunctional phytoene synthase / lycopene cyclase, and the crtI gene which codes for carotene desaturase. Note that crtB and crtY are usually separate enzymes: phytoene synthase and lycopene cyclase respectively. crtYB is essentially crtB with an active domain from crtY, allowing it to catalyze both these two non-sequential reactions, in addition to a third reaction which converts neurosporene into dihydro-beta-carotene, resulting in a loss of carbon from this pathway.
| + | ====== Objectives ====== |
| | + | * Compare the various modeling languages and platforms available |
| | + | * Use our choice modeling language to develop a generalizable BioBrick model |
| | | | |
| - | Farnesyl diphosphate is the first metabolite that we consider in our synthetic pathway because it is the last metabolite found normally in yeast. We list our other metabolites in their order in the pathway until our final product beta-carotene. We conclude with a list of enzymatic kinetic parameters (K<sub>m</sub> and K<sub>cat</sub> per Michaelis-Menten) and a list of expression kinetic parameters. These lengthy rate declarations are not shown below, but can be seen in the full code.
| + | ====== A BioBrick in Kappa ====== |
| | + | BioBricks are meant to be reusable and composable. While the BioBrick sequences may follow these principles, models of BioBrick-based systems often do not. Ty Thomson's Kappa model is a good example of an attempt to standardize BioBrick model<sup>[[#Foot1|[1]]]</sup>. Thomson's model is designed for use in E. coli, which alone limits its power and would force us to redesign it in our application. However the model suffers from more severe and general drawbacks. In order to take advantage of Kappa's rule-based engine, Thomson creates a general DNA agent with upstream, downstream, and binding sites. Thus any rules applied to DNA will apply to all BioBricks. However, often you want rules to refer to a specific BioBrick. Different BioBricks make different proteins, will be transcribed at different rates, or bind certain repressors. Thomson gets around this problem with a hack: he creates a dummy binding site called "label" and gives it a state such as "BBa000011" to identify the BioBrick. When, for example, a modeler wishes to specify unique transcription rates for each BioBrick, he or she is forced to write the same reaction for each BioBrick, changing only the rate and the state of the label "binding site". While functional, the unattractive nature of this hack points to a fundamental problem in Kappa. |
| | | | |
| - | <pre>
| + | ====== A BioBrick in LBS ====== |
| - | // Compartments
| + | Enter LBS's module facility. Modules allow the modeler to ''the structure of a complex interaction once'', and paramaterize that structure with rates, compartments, and agents. Consider the extremely simple gene expression model below adapted from Pedersen's thesis<sup>[[#Foot2|[2]]]. |
| - | comp cytosol = new comp;
| + | |
| - | comp nucleus = new comp inside cytosol;
| + | |
| | | | |
| - | // Transcriptional machinery
| + | <pre> |
| - | spec Ribo = new{mrna:binding};
| + | module m(comp nuc; rate trnsc; spec gene, prot){ |
| - | spec RNAP = new{dna:binding,mrna:binding}; | + | spec mrna = new{}; |
| - | // Gene-protein pairs
| + | nuc[gene + rnap ->{transc} gene + rnap + mrna] | |
| - | spec crtE = new{bind:binding}; spec GGPP_synth_exo = new{};
| + | nuc[mrna] ->{1} mrna | |
| - | spec BTS1 = new{bind:binding}; spec GGPP_synth_endo = new{}; | + | rs + mrna ->{0.1} rs +prot |
| - | spec crtYB = new{bind:binding}; spec carotene_desat = new{};
| + | }; |
| - | spec crtI = new{bind:binding}; spec phytoene_synth_lycopene_cyc = new{};
| + | |
| - | // Metabolites
| + | |
| - | spec farnesyl_PP = new{};
| + | |
| - | spec GGPP = new{};
| + | |
| - | spec phytoene = new{};
| + | |
| - | spec neurosporene = new{};
| + | |
| - | spec lycopene = new{};
| + | |
| - | spec beta_carotene = new{};
| + | |
| - | spec hh_beta_carotene = new{};
| + | |
| | </pre> | | </pre> |
| | | | |
| - | ==== Expression ==== | + | Adding the line m(nucleus, .001, crtI, phytoene_desaturase) will cause gene expression at a specific rate to occur. Unlike in Kappa, this required no "dummy" labeling and no chemical reaction had to be declared. Thus '''the model's abstract structure is totally separated from the realization of that structure for specific systems'''. This module can be reused precisely as it is to model another system. Our BioBrick expression model in LBS makes extensive use of LBS's powerful module facility. |
| - | The first three modules comprise [https://2011.igem.org/Team:Johns_Hopkins/Modeling/GeneExp our standard expression model].
| + | |
| | + | The following schematic depicts the model we developed in LBS. It was generated in part by [http://lepton.research.microsoft.com/webgec/ Visual GEC], the software environment used to compile and run LBS simulations. |
| | + | |
| | + | [[File:Expression.jpg|610px|BioBrick expression module implemented in LBS]] |
| | + | |
| | + | ===== Expression module ===== |
| | + | We interpret gene expression as a process parameterized by a specific gene and by the specific protein to be made. We also require a compartment to serve as the nucleus, since expression in yeast needs to be compartment-aware. While we take the DNA and protein as given, we declare mRNA in the scope of the expression module. Each gene-protein pair will now get a unique mRNA that is abstracted away from all other interactions in the model. We pass off the work to two sub-modules: transcribe and translate. These modules need not themselves be aware of multiple compartments, so we write a line specifying nuclear export of the mRNA. We also perform RNA degradation in the nucleus in parallel with transcription and in the cytosol in parallel with translation. |
| | <pre> | | <pre> |
| - | module transcribe(spec DNA:{bind}, mRNA:{down}) {...}; | + | module express(comp nuc; spec DNA:{bind}, Prot) { |
| - | module translate (spec mRNA:{up}, Prot) {...};
| + | spec mRNA = new{up:binding,down:binding}; |
| - | module express (comp nuc; spec DNA:{bind}, Prot) {...};
| + | nuc[ transcribe(DNA:{bind}, mRNA:{down}) | mRNA ->{rna_deg} ] | |
| | + | nuc[ mRNA ] ->{export} mRNA | |
| | + | translate(mRNA:{up}, Prot) | mRNA -> {rna_deg} |
| | + | }; |
| | </pre> | | </pre> |
| - | | + | ===== Transcription module ===== |
| - | ==== Simple reaction module ==== | + | Transcription requires a DNA segment and an mRNA product. In our model, RNA polymerase, which was declared globally, binds DNA. Then the mRNA molecule appears also bound to RNA polymerase. Finally, the three molecules dissociate. These reactions are irreversible. |
| - | We create a module to handle simple reactions in which an enzyme converts a single substrate to a single product. All metabolic reactions that we are interested in follow the Michaelis-Menten pattern outlined below. Note that the rate here is not a rate constant as it would be in mass action kinetics, but is instead the Michaelis-Menten rate expression.
| + | |
| | <pre> | | <pre> |
| - | module mmRXN(spec sub, enz, prod; rate km, kcat) { | + | module transcribe(spec DNA:{bind}, mRNA:{down}) { |
| - | rate rxn_rate = kcat*enz/(sub+km); | + | RNAP{dna} + DNA{bind} ->{rnap_bind} RNAP{dna!1}-DNA{bind!1} | |
| - | sub + enz ->[rxn_rate] enz + prod | + | RNAP{dna!1}-DNA{bind!1} ->{transcription} mRNA{down!2}- |
| | + | RNAP{dna!1,mrna!2}-DNA{bind!1} | |
| | + | mRNA{down!2}-RNAP{dna!1,mrna!2}-DNA{bind!1} ->{termination} |
| | + | mRNA{down} + RNAP{dna,mrna} + |
| | + | DNA{bind} |
| | }; | | }; |
| | </pre> | | </pre> |
| | | | |
| - | ==== Expression and reaction module invocations ==== | + | ===== Translation module ===== |
| - | This is the heart of the code where we tell LBS to actually simulate something. We invoke the expression module on our two gene-enzyme pairs and the reaction modules for the metabolites that our enzymes process. We then explicitly degrade our enzymes at a uniform rate. The vertical pipes are parallel operators which tell LBS to evaluate these reactions in parallel. In the actual code, all the code below is encapsulated in cytosol[...].
| + | The translation module is similar to and even simpler than transcription. mRNA binds a ribosome (declared globally) in a reversible fashion. Once bound, the ribosome can generate a protein and release both the protein and mRNA in a single step. |
| - | | + | |
| - | It is important to note that our model greatly oversimplifies the complexity of the reactions. Each conversion between metabolites involves between 2 and 4 separate reactions. However, these reactions tend to be very fast. While the enzymes in our pathway are considered to catalyze all steps of these reactions, the overall rate is controlled by the enzyme's ability to catalyze a single rate-limiting reaction. In fact, the other minor reaction rates are often not even measurable. You will note one exception below in the case of carotene desaturase. Carotene desaturase converts phytoene to neurosporene at a rate of about 120 per second. It then converts neurosporene to lycopene at a rate of about 10 per second. Even though we model both reactions explicitly, the observation that one reaction is an order of magnitude slower than the other supports the single rate-limiting step simplification.
| + | |
| | <pre> | | <pre> |
| - | express(nucleus, crtE:{bind}, GGPP_synth_exo) |
| + | module translate(spec mRNA:{up}, Prot) { |
| - | express(nucleus, BTS1:{bind}, GGPP_synth_endo) |
| + | Ribo{mrna} + mRNA{up} <->{ribo_bind}{ribo_unbind} |
| - | express(nucleus, crtYB:{bind}, phytoene_synth_lycopene_cyc) |
| + | Ribo{mrna!1}-mRNA{up!1} | |
| - | express(nucleus, crtI:{bind}, carotene_desat) |
| + | Ribo{mrna!1}-mRNA{up!1} ->{translation} Ribo{mrna} + mRNA{up} + Prot |
| - | mmRXN(farnesyl_PP, GGPP_synth_endo, GGPP, GGPP_synth_endo_Km, GGPP_synth_endo_Kcat) |
| + | }; |
| - | mmRXN(farnesyl_PP, GGPP_synth_exo, GGPP, GGPP_synth_exo_Km, GGPP_synth_exo_Kcat) |
| + | |
| - | mmRXN(GGPP, phytoene_synth_lycopene_cyc, phytoene, phytoene_synth_Km, phytoene_synth_Kcat) |
| + | |
| - | mmRXN(phytoene, carotene_desat, neurosporene, carotene_desat_neurosporene_Km,
| + | |
| - | carotene_desat_neurosporene_Kcat) |
| + | |
| - | mmRXN(neurosporene, carotene_desat, lycopene, carotene_desat_lycopene_Km,
| + | |
| - | carotene_desat_lycopene_Kcat) |
| + | |
| - | mmRXN(neurosporene, carotene_desat, hh_beta_carotene,
| + | |
| - | phytoene_synth_hh_beta_carotene_Km, phytoene_synth_hh_beta_carotene_Kcat) |
| + | |
| - | mmRXN(lycopene, phytoene_synth_lycopene_cyc, beta_carotene,
| + | |
| - | phytoene_synth_beta_carotene_Km, phytoene_synth_beta_carotene_Kcat) |
| + | |
| - | GGPP_synth_exo ->{prot_deg} |
| + | |
| - | GGPP_synth_endo ->{prot_deg} |
| + | |
| - | carotene_desat ->{prot_deg} |
| + | |
| - | phytoene_synth_lycopene_cyc ->{prot_deg} |
| + | |
| | </pre> | | </pre> |
| | | | |
| - | ==== Initial conditions ====
| + | Parameters for this model can be found [https://2011.igem.org/Team:Johns_Hopkins/Modeling/Para here]. |
| - | We establish initial conditions for our system, namely 5 copies of each gene and a very limited number of ribosomes and RNA polymerases. We take the numbers here to mean "the number of ribosomes or RNA polymerases made available to the expression process of our genes of interest". There are of course many times more ribosomes and polymerases in the cell, but they are working on making other proteins. Only a small portion is ever free to deal with our synthetic construct.
| + | |
| | | | |
| - | In addition, the first line below is a black-box input: a certain number of our initial substrate is dumped into our system each turn by the cell. We are not interested in modeling the process of how it comes about, we only care about the result and what our system does to this exogenous input.
| + | ====== A Simplified Expression Model ====== |
| - | <pre> | + | The expression model above requires the number of ribosomes and polymerases available to our system, and the various rate constants associated with them, to be estimated. We can measure the final quantity of protein produced and the number of copies of the gene introduced into the cell, but this information is insufficient to estimate all the parameters in the model. Further, literature values regarding these values are sparse, crude, and often contradictory. We decided to build a simplified model with fewer parameters. |
| - | ->{1} farnesyl_PP |
| + | |
| - | init Ribo 10 |
| + | [[File:Simple_expression.jpg|610px|Simplified expression model]] |
| - | nucleus[
| + | |
| - | init crtE 5 |
| + | This system is so simplistic that it is quite easy to solve for the steady-state concentrations of all species analytically. In fact, we find that if |
| - | init crtYB 5 |
| + | t<sub>s</sub> = transcription rate |
| - | init crtI 5 |
| + | t<sub>l</sub> = translation rate |
| - | init RNAP 1
| + | p = protein degradation rate |
| - | ] | + | r = mRNA degradation rate |
| - | </pre> | + | c = number of gene copies in the cell |
| | + | |
| | + | \[[Protein]=(t_{s}\cdot t_{l})/(p\cdot r)\cdot c\] |
| | + | |
| | + | This is useful when translating our pathway optimization results into an implementation strategy for real organisms. We perform our optimization on Matlab models that do not include gene expression. Thus the optimal parameters are stated in terms of enzyme concentrations. Given the target enzyme concentrations suggested by our optimization algorithm, an accurate enough expression model could then allow us to infer optimal gene copy numbers and promoter strengths. We can then search a database of yeast promoter strengths to select the promoter that should be used and the gene copy number needed. In practice this is unrealistic. Predictive models of mRNA lifetime do not exist. Transcription and translation rates are fairly sensitive to the specific codons used. These processes are also exceptionally noisy. It turns out that the metabolic kinetics are much easier to describe accurately, so our results from [https://2011.igem.org/Team:Johns_Hopkins/Modeling/Opt optimization] are useful. However, inferring the genetic construct needed to achieve the optimal protein concentrations will require trial-and-error at the bench. |
| | + | |
| | + | ======References====== |
| | + | <span id="Foot1"><sup>[1]</sup>Ty Thompson. Rule-Based Modeling of BioBrick Parts. http://www.rulebase.org/books/184351-Rule-Based-Modeling-of-BioBrick-Parts.</span> |
| | + | |
| | + | <span id="Foot2"><sup>[2]</sup> Pedersen, M., & Plotkin, G. D. (2010). A language for biochemical systems: Design and formal specification. (C. Priami, R. Breitling, D. Gilbert, M. Heiner, & A. M. Uhrmacher, Eds.)Transactions on Computational Systems Biology XII, 5945, 77-145. Springer Berlin Heidelberg.</span> |
| | | | |
| | | | |
| - | <html> | + | <html></div> |
| - | </div> | + | |
Introduction
Once we sketched out schematic graphical models of vitamin A and vitamin C production, we set out to find the perfect modeling software. We wanted something simple, expressive, powerful, and extendable. We started where last year's modeling prize winner left off: [http://kappalanguage.org/ Kappa].
Objectives
- Compare the various modeling languages and platforms available
- Use our choice modeling language to develop a generalizable BioBrick model
A BioBrick in Kappa
BioBricks are meant to be reusable and composable. While the BioBrick sequences may follow these principles, models of BioBrick-based systems often do not. Ty Thomson's Kappa model is a good example of an attempt to standardize BioBrick model[1]. Thomson's model is designed for use in E. coli, which alone limits its power and would force us to redesign it in our application. However the model suffers from more severe and general drawbacks. In order to take advantage of Kappa's rule-based engine, Thomson creates a general DNA agent with upstream, downstream, and binding sites. Thus any rules applied to DNA will apply to all BioBricks. However, often you want rules to refer to a specific BioBrick. Different BioBricks make different proteins, will be transcribed at different rates, or bind certain repressors. Thomson gets around this problem with a hack: he creates a dummy binding site called "label" and gives it a state such as "BBa000011" to identify the BioBrick. When, for example, a modeler wishes to specify unique transcription rates for each BioBrick, he or she is forced to write the same reaction for each BioBrick, changing only the rate and the state of the label "binding site". While functional, the unattractive nature of this hack points to a fundamental problem in Kappa.
A BioBrick in LBS
Enter LBS's module facility. Modules allow the modeler to the structure of a complex interaction once, and paramaterize that structure with rates, compartments, and agents. Consider the extremely simple gene expression model below adapted from Pedersen's thesis[2].
module m(comp nuc; rate trnsc; spec gene, prot){
spec mrna = new{};
nuc[gene + rnap ->{transc} gene + rnap + mrna] |
nuc[mrna] ->{1} mrna |
rs + mrna ->{0.1} rs +prot
};
Adding the line m(nucleus, .001, crtI, phytoene_desaturase) will cause gene expression at a specific rate to occur. Unlike in Kappa, this required no "dummy" labeling and no chemical reaction had to be declared. Thus the model's abstract structure is totally separated from the realization of that structure for specific systems. This module can be reused precisely as it is to model another system. Our BioBrick expression model in LBS makes extensive use of LBS's powerful module facility.
The following schematic depicts the model we developed in LBS. It was generated in part by [http://lepton.research.microsoft.com/webgec/ Visual GEC], the software environment used to compile and run LBS simulations.
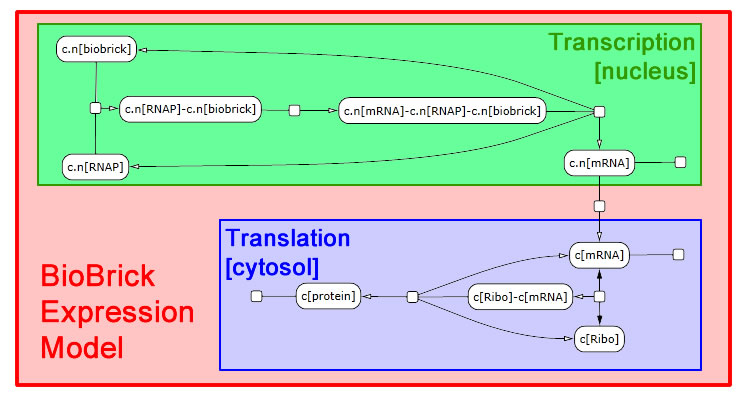
Expression module
We interpret gene expression as a process parameterized by a specific gene and by the specific protein to be made. We also require a compartment to serve as the nucleus, since expression in yeast needs to be compartment-aware. While we take the DNA and protein as given, we declare mRNA in the scope of the expression module. Each gene-protein pair will now get a unique mRNA that is abstracted away from all other interactions in the model. We pass off the work to two sub-modules: transcribe and translate. These modules need not themselves be aware of multiple compartments, so we write a line specifying nuclear export of the mRNA. We also perform RNA degradation in the nucleus in parallel with transcription and in the cytosol in parallel with translation.
module express(comp nuc; spec DNA:{bind}, Prot) {
spec mRNA = new{up:binding,down:binding};
nuc[ transcribe(DNA:{bind}, mRNA:{down}) | mRNA ->{rna_deg} ] |
nuc[ mRNA ] ->{export} mRNA |
translate(mRNA:{up}, Prot) | mRNA -> {rna_deg}
};
Transcription module
Transcription requires a DNA segment and an mRNA product. In our model, RNA polymerase, which was declared globally, binds DNA. Then the mRNA molecule appears also bound to RNA polymerase. Finally, the three molecules dissociate. These reactions are irreversible.
module transcribe(spec DNA:{bind}, mRNA:{down}) {
RNAP{dna} + DNA{bind} ->{rnap_bind} RNAP{dna!1}-DNA{bind!1} |
RNAP{dna!1}-DNA{bind!1} ->{transcription} mRNA{down!2}-
RNAP{dna!1,mrna!2}-DNA{bind!1} |
mRNA{down!2}-RNAP{dna!1,mrna!2}-DNA{bind!1} ->{termination}
mRNA{down} + RNAP{dna,mrna} +
DNA{bind}
};
Translation module
The translation module is similar to and even simpler than transcription. mRNA binds a ribosome (declared globally) in a reversible fashion. Once bound, the ribosome can generate a protein and release both the protein and mRNA in a single step.
module translate(spec mRNA:{up}, Prot) {
Ribo{mrna} + mRNA{up} <->{ribo_bind}{ribo_unbind}
Ribo{mrna!1}-mRNA{up!1} |
Ribo{mrna!1}-mRNA{up!1} ->{translation} Ribo{mrna} + mRNA{up} + Prot
};
Parameters for this model can be found here.
A Simplified Expression Model
The expression model above requires the number of ribosomes and polymerases available to our system, and the various rate constants associated with them, to be estimated. We can measure the final quantity of protein produced and the number of copies of the gene introduced into the cell, but this information is insufficient to estimate all the parameters in the model. Further, literature values regarding these values are sparse, crude, and often contradictory. We decided to build a simplified model with fewer parameters.

This system is so simplistic that it is quite easy to solve for the steady-state concentrations of all species analytically. In fact, we find that if
ts = transcription rate
tl = translation rate
p = protein degradation rate
r = mRNA degradation rate
c = number of gene copies in the cell
\[[Protein]=(t_{s}\cdot t_{l})/(p\cdot r)\cdot c\]
This is useful when translating our pathway optimization results into an implementation strategy for real organisms. We perform our optimization on Matlab models that do not include gene expression. Thus the optimal parameters are stated in terms of enzyme concentrations. Given the target enzyme concentrations suggested by our optimization algorithm, an accurate enough expression model could then allow us to infer optimal gene copy numbers and promoter strengths. We can then search a database of yeast promoter strengths to select the promoter that should be used and the gene copy number needed. In practice this is unrealistic. Predictive models of mRNA lifetime do not exist. Transcription and translation rates are fairly sensitive to the specific codons used. These processes are also exceptionally noisy. It turns out that the metabolic kinetics are much easier to describe accurately, so our results from optimization are useful. However, inferring the genetic construct needed to achieve the optimal protein concentrations will require trial-and-error at the bench.
References
 "
"


