 "
"

Violacein loxPsym Integration
6/11/2011
Verify that the promoters and 3'UTRs are present in the plasmid library made by Yuxuan. The inserts are ligated into pCR 2.1-TOPO and contain BsaI sites, so following digestion with BsaI they should be visible on a gel.
Note: Due to a lack of good sequences the promoters and 3'UTRs for RPL8A, FCY2, and KRE9 were not available.
Minipreps were prepared by Ashan.
20µl digest
| Component | Volume (µl) |
| Plasmid DNA | 5 |
| 10x NEBuffer 4 | 2 |
| H2O | 12.5 |
| BsaI (10U/l) | .5 |
| Total | 20 |
~2.5 hour incubation @ 50°C.
The 3'UTRs were expected to be small (most ~142bp), so they were run on a 2% MetaPhor agarose gel.
.jpg)
Results:
None of the expected bands were observed. The bands at ~1kb are pieces of the vector (pCR2.1-TOPO), which contains a BsaI site in its sequence.
6/12/2011
Gel electrophoresis of promoters from 6/11 restriction digest to verify presence in provided plasmids.
Results:

Some bands are visible around 500bp, but they are not consistent with the expected size of the promoters. The ladder is also very weak.
6/20/2011
The 6/11 digest was run with BsaI at 50°C based on Dr. Boeke's NEB book, but the NEB website recommends 37°C. I will run a digest on lambda DNA provided by Dr. Boeke to test which temperature yields better results.
Was given 10µl of DNA, 1µg/µl. Made two master mixes in order to make a titration of BsaI, enough for 7 digests each.
| Reaction Mix 1 | x7 | Master Mix 1 | Reaction Mix 2 | x7 | Master Mix 2 | |
| .67µl DNA | 4.67µl DNA | .67µl DNA | 4.67µl DNA | |||
| 17.13µl H2O | 119.91µl H2O | 17.33µl H2O | 121.31µl H2O | |||
| 2µl 10x B4 | 14µl 10x B4 | 2µl 10x B4 | 14µl 10x B4 | |||
| .2µl BsaI | 1.4µl BsaI | |||||
| 20µl total, 3 units Bsa1/DNA | 140µl total | 20µl total, 0 units BsaI/DNA | 140µl total |
| Titration | ||||
| 3x | 2x | 1x | 1/3x | 0x (-control) |
| 60µl MM1 | 40µl MM1 | 20µl MM1 | 6.67µl MM1 | |
| 20µl MM2 | 40µl MM2 | 53.33µl MM2 | 60µl MM2 |

Digestion at 50°C yielded slightly clearer bands. This means that the promoter/UTR digestion was performed at the correct temperature, and the digestion temperature does not explain the unexpected results. Additionally, it seems that the 2-log ladder with CS red that we had been using (the leftmost column) is showing up very faintly, so we will stop using it.
6/22/2011
The unexpected digestion patterns might be explained if the plate with the plasmids sent out for sequencing was flipped. I will do colony PCR on the sequencing plates to test this hypothesis. If this fails, I will PCR the promoters and UTRs from genomic DNA.
I chose 6 promoters of varying sizes to test. The promoters are generally larger than the UTRs so they should be more visible on a gel.
| Label | Promoter | Length (bp) | position | flipped position | Label | Fragment | Length (bp) |
| 1 | THD3 | 534 | A2 | H11 | 1F | BAP2utr | 542 |
| 2 | RPL24A | 664 | A9 | H4 | 2F | HHO1utr | 354 |
| 3 | RPS8B | 834 | B7 | G5 | 3F | KRE9utr w/mutation | 263 |
| 4 | STM1 | 534 | D7 | E6 | 4F | FCY2utr w/mutation | 142 |
| 5 | ARD1 | 535 | F5 | E9 | 5F | RPS2utr | 142 |
| 6 | BAP2 | 834 | H5 | F9 | 6F | TDH3utr | 142 |
The E. coli containing these plasmids were streaked out on LB and incubated at 37°C, along with the strains containing STM1, ARD1, and BAP2 the original restriction digests were performed on.
6/24/2011
The bacteria were left in the incubator for two days, but I decided to proceed since the PCR did not need to select for single colonies.

Did four reactions for each promoter, flipped sequence, and original sequence.
No bands were observed. Too many cells may have been added to the reaction, inhibiting the PCR. Also, the primers were 5µM rather than the 5mM written on the plate, so not enough primer was used.
6/27/2011
The 6/24 colony PCR failed, so I will repeat the PCR, adding fewer cells to the reaction, using the correct concentration of primer, and extending the initial denaturation step.
Followed same protocol as 6/24, trying not to pick too many cells, using .15µl of the 5µM primer mix, and extending the initial denaturation to 10 minutes. PCR product was stored @ 4°C.
6/30/2011
Ran the 6/27 PCR product.

No bands observed. It was probably a bad idea modifying a yeast protocol for bacteria, will use a more suitable protocol and try again.
7/1/2011
Still trying to get a colony PCR to work in order to verify the promoters and UTRs are present, this time with a different protocol. Tested 6 promoters and their corresponding 'flipped' positions on the sequencing place in order to test the hypothesis that the sequencing plate was flipped.
Followed Marty's protocol for colony PCR using GoTaq Green.

Still nothing observed besides primers.
7/5/2011
One more attempt at the colony PCR. Same as 7/1 procedure.

Same pattern of only primers/primer dimers/partial products observed 7/1. Move to PCR the promoters and 3'UTRs from genomic DNA instead.
7/7/2011
Test whether the gDNA PCR will work, using a small set of the promoters and 3'UTRs.
Received two sets of wild-type genomic DNA from Zheng, one fully processed and one semi-processed, along with a positive control primer pair (12336/9613) and a negative control primer pair (12310/9613). Because the control primers were 20µM and the promoter/UTR primers were 5µM, made two reaction mixes.
Promoter/UTR gDNA PCR Reaction Mix
| Component | Volume |
| gDNA | 2 µl |
| 10x ExTaq buffer | 2 µl |
| dNTP mix (2.5mM ea.) | 1 µl |
| F/R Primer mix (5µM) | 3.2 µl |
| ExTaq | .1 µl |
| dH2O | 11.7 µl |
| Total | 20 µl |
Control Reaction Mix
| Component | Volume |
| gDNA | 2 µl |
| 10x ExTaq buffer | 2 µl |
| dNTP mix (2.5mM ea.) | 1 µl |
| F primer (20µM) | .4 µl |
| R primer (20µM) | .4 µl |
| ExTaq | .1 µl |
| dH2O | 14.1 µl |
| Total | 20 µl |
PCR Program: 5 min @ 94°C, 30x(30s @ 94°C, 30s @ 55°C, 30s @ 72°C), 5 min @ 72°C, hold @ 4°C

All the bands showed up as expected with the fully processed gDNA.
7/8/2011
Should have nanodropped the gDNA to see how much to put in 7/7 reaction, so did that.
gDNA: 63.2 ng/µl
7/12/2011
PCR out all of the promoters/3'UTRs w/BsaI compatible primers for later ligation with ORFs and loxPsym site.
50µl reaction volume and Herculase polymerase. Got two positive primers from Zheng (ZK096/097, ZK098/099).
Promoter/UTR gDNA PCR Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (63ng/µl) | 2 µl |
| F/R Primer mix (5µM) | 5 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 27.5 µl |
| Total | 50 µl |
Control Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (63ng/µl) | 2 µl |
| F Primer (20µM) | .625 µl |
| R Primer (20µM) | .625 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 31.25 µl |
| Total | 50 µl |
PCR Program: 2 min @ 95°C, 30x(20s @ 95°C, 20s @ 56°C, 30s @72°C), 3 min @ 72°C, hold at 4°C.
Took 10µl for gel to check for product.

Will PCR purify.
7/13/2011
PCR purification of 7/12 PCR product using Qiagen kit. Ran 10µl to check products.

7/15/2011
Based on the 7/13 PCR purification, I need to gel extract, because there is lots of junk at around 100bp for many of the reactions. I will do a new PCR with reduced primer concentration.
Got gDNA from Patrick, 138ng/µl. 50µl reaction using Herculase.
Promoter/UTR Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (138ng/µl) | 1 µl |
| F/R Primer mix (5µM) | 1 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 32.5 µl |
| Total | 50 µl |
Control Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (63ng/µl) | 1 µl |
| F Primer (20µM) | .125 µl |
| R Primer (20µM) | .125 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 33.25 µl |
| Total | 50 µl |
PCR Program: 2 min @ 95°C, 30x(20s @ 95°C, 20s @ 56°C, 30s @72°C), 3 min @ 72°C, hold at 4°C.


Not enough DNA for extraction, will adjust primer concentration again.
7/18/2011
Promoter/UTR gDNA PCR attempt 3, now with an intermediate primer concentration.
Procedure:
Promoter/UTR Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (138ng/µl) | 1 µl |
| F/R Primer mix (5µM) | 2.5 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 31 µl |
| Total | 50 µl |
Control Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (63ng/µl) | 1 µl |
| F Primer (20µM) | .3125 µl |
| R Primer (20µM) | .3125 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 32.875 µl |
| Total | 50 µl |
PCR Program: 2 min @ 95°C, 30x(20s @ 95°C, 20s @ 56°C, 30s @72°C), 3 min @ 72°C, hold at 4°C.
7/19/2011
Purified DNA from 7/18 gel extraction according to gel extraction protocol, then nanodropped to find concentration. Used Invitrogen spin columns I found in the columns box.
| Sequence | Concentration (ng/µl) | Sequence | Concentration (ng/µl) |
| TDH3p | 31.6 | FCY2p | 31.7 |
| TDH3utr | 13.7 | FCY2utr | 22.9 |
| RPL24Ap | 37.9 | PRY1p | 4.6 |
| RPL24Autr | 12.6 | PRY1utr | 21.9 |
| RPS8Bp | 22.2 | ARD1p | 3.0 |
| RPS8Butr | 5.1 | ARD1utr | 9.7 |
| RPS2p | 2.9 | KRE9p | 42.1 |
| RPS2utr | 3.7 | KRE9utr | 41.0 |
| RPL8Ap | 3.0 | HHO1p | 49.4 |
| RPL8Autr | 18.6 | HHO1utr | 14.6 |
| STM1p | 31.5 | BAP2p | 9.3 |
| STM1utr | 17.9 | BAP2utr | 27.9 |
7/21/2011
Loaded 5µl of product on a 1% gel to verify 7/19 gel purification products.

7/25/11
Colony purified ß-carotene yeast strains in preparation for gDNA PCR experiment investigating white/super orange colonies. Inoculated YPD and dough media plates w/Rob for ß-carotene assay. Prepared WT gDNA for Dan W.
7/26/11
Try to PCR missing promoter and 3'UTRs from gDNA using adjusted annealing temperature.
Promoter/UTR Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (138ng/µl) | 1 µl |
| F/R Primer mix (5µM) | 5 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 28.5 µl |
| Total | 50 µl |
Control Reaction Mix
| Component | Volume |
| 5x Herc. buffer | 10 µl |
| dNTP mix (2.5mM ea.) | 5 µl |
| gDNA (63ng/µl) | 1 µl |
| F Primer (20µM) | .625 µl |
| R Primer (20µM) | .625 µl |
| Herculase Polymerase | .5 µl |
| dH2O | 32.25 µl |
| Total | 50 µl |
PCR Program: 4 min @ 95°C, 30x(30s @ 95°C, 30s @ 58°C, 30s @72°C), 7 min @ 72°C, hold at 4°C.
Also ran 3 timepoints of ß-carotene assay w/Rob.
7/27/2011
Gel extraction of 7/26 PCR products. Cut gels and took mass for purification, stored in 4°C overnight.
7/28/2011
Finish purification of 7/26 PCR product according to gel extraction with microcentrifuge, ran 5µl on gel to check.

| Sequence | Concentration (ng/µl) | Sequence | Concentration (ng/µl) | |
| TDH3p | 38.8 | FCY2p | 41.8 | |
| TDH3utr | 21.3 | FCY2utr | 26.1 | |
| RPL24Ap | 53.1 | PRY1p | 14.9 | |
| RPL24Autr | 26.4 | PRY1utr | 41.2 | |
| RPS8Bp | 40.9 | ARD1p | 4.4 | |
| RPS8Butr | 9.7 | ARD1utr | 21.1 | |
| RPS2p | 3.4 | KRE9p | 66.3 | |
| RPS2utr | 16.5 | KRE9utr | 54.3 | |
| RPL8Ap | 3.2 | HHO1p | 48.5 | |
| RPL8Autr | 21.8 | HHO1utr | 15.2 | |
| STM1p | 43.0 | BAP2p | 8.4 | |
| STM1utr | 31.3 | BAP2utr | 34.3 |
7/29/2011
Linearize vector pTZ19u w/SmaI in order to blunt-end ligate the BsaI containing fragments in for cloning and attaching URA3 to UTRs.
20µl digest
| Component | Volume (µl) |
| ptz19u (60ng/µl) | 20 |
| NEBuffer 4 (10x) | 5 |
| BSA (100x) | .5 |
| SmaI | 1 |
| H2O | 23.5 |
| Total | 50 |
Left to digest overnight at room temperature (~23°C), rather than suggested temperature 25°C.
7/30/2011
Heat-killed SmaI (20 min @ 65°C).
Diluted pTZ19u 10x to 156.4 ng/µl (50µl total).
Ran gel of digested and undigested pTZ19u.

SmaI digest looks good; will go ahead with ligation.
Ligation Mix (20µl)
| Component | Volume |
| 10x T4 Buffer | 2µl |
| SmaI linearized pTZ19u (58ng/µl) | 1µl |
| T4 ligase (120,000 U/ml) | 1µl |
| insert | 6:1 molar ratio insert:vector |
| H2O | to 20µl |
Left in 16°C overnight.
8/1/2011
Streaked out TOP10 e. coli on LB+carb for making competent cells.
8/2/2011
The TOP10 plates were accidentally placed in 30°C incubator instead of 37°C incubator overnight, there was no growth at all. Also I think you have to streak out on plain LB or the carb will kill the cells as they come out of -80°C.
Streaked out two new plates of TOP10 cells, one on LB, one on LB+carb, put in 37°C incubator along with the 8/1 plate.
Came back in evening, some growth on LB only plate, but no colonies for inoculation yet, so will have to delay one day.
8/3/2011
Inoculated 25ml LB with TOP10 E. coli, put in 37°C w/shaking.
Made gDNA from orange yeasts according to this protocol upload and link to protocol here. Accidentally forgot to spin and discard supernatant before adding glass beads, so spun 5 min @ 14000 rpm after adding beads and then discarded supernatant. Got pretty low concentrations:
| Strain | gDNA concentration (ng/µl) |
| 039 | 54.6 |
| 179 | 85.9 |
| 201 | 85.1 |
| 128 | 33.4 |
| 128 Super Orange | 81.1 |
| 128 White | 63.1 |
8/4/2011
Prepared competent cells according to competent E. coli preparation protocol.
There were no LB Carb plates for transformations of the 7/30 ligation, so spread 90µl of an X-Gal/IPTG/Carb mix on plain LB plates.
| Component | Volume (µl) |
| X-Gal (1000x) | 25 |
| 100mM IPTG | 40 |
| Carbenicillin (1000x) | 25 |
| Total | 90 |
Followed Top10 transformation protocol for all promoters and UTRs except for RPS2utr (from 7/30 ligation). Spread one set of 50µl cells/ 35µl LB and one set of 200µl cells/ 85µl LB. Placed in 37°C overnight.
8/5/2011
Blue/white colonies observed for both 50µl and 200µl plates, but were very small, so incubated further. Spread 25µl 1000x Carbenicillin on plain LB plates to streak out colonies. Picked one white colony for each promoter/UTR and restreaked; put in 37°C incubator. Blue/white plates moved to iGEM 4°C space.
8/6/2011
Inoculated promoter/UTR E. coli into 5ml LB. Moved 8/5 plates to iGEM 4°C space.
8/8/2011
All overnight cultures looked ok except for PRY1p, which was yellowish. Perhaps contamination?
Miniprepped all promoter/UTR overnights according to protocol.
Performed restriction digest with BsaI. If the ligation/transformation was successful, the promoter/UTR should be cut out of the vector.



Made 15% glycerol stocks of E. coli w/correct fragments present, put in iGEM -80°C space.
8/9/2011
PCR of URA3 to get product and add NotI site to add to 3' UTR.
Diluted ura3_fwd and ura3_rev primers to 10µM (50µl 60µM primer, 250µl TE). 50µl reaction using Herculase II Polymerase.
| Component | Volume (µl) |
| 5x Herculase Buffer | 10 |
| dNTP mix (2.5mM ea.) | 5 |
| gDNA (148.8ng/µl) | .67 (100ng), 1.34 (200ng), 2.01 (300ng) |
| forward primer (10µM) | 1.25 |
| reverse primer (10µM) | 1.25 |
| dH2O | to 50 |
| Total | 50 |
Control primers were 20µM (ZK096/097), used .625µl ea.
PCR Program: 2 min @ 95°C, 30x (20s @95°C, 20s @64°C, 34s @72°C), 3 min @ 72°C
Ran gel under long wave in preparation for extraction, only saw faint product for 300ng band, decided not enough DNA to cut.
8/10/2011
Looking at 8/8 gels, need to redo ligation and transformation for TDH3utr, RPS8Butr, RPS2utr, RPL8Ap, RPL8Autr, STM1utr, FCY2utr, PRY1p, PRY1utr, ARD1utr, KRE9utr, HH01p, HH01utr, BAP2utr.
Linearized pTZ19u w/SmaI.
50µl reaction
| Component | Volume (µl) |
| pTZ19U (156.4 ng/µl) | 7.67 |
| 10x NEBuffer 4 | 5 |
| BSA (100x) | .5 |
| SmaI (20U/µl) | 1 |
| dH2O | 35.83 |
| Total | 50 |
2 hour digest, heat kill SmaI 65°C 20 min.
20µl ligation mix
| Component | Volume (µl) |
| 10x T4 buffer | 2 |
| T4 ligase (Enzymatics: 120U/µl) | 1 |
| linearized pTZ19u (58ng/µl) | 1 |
| insert | 6x molar ratio insert/vector |
| dH2O | to 20 |
| Total | 20 |
Moved to 25°C for overnight.
Redid URA3 PCR. Trying primers suspended in H2O as well as in TE, and lowering the annealing temperature to 61°C vs 64°C.
Both primer sets yielded a good product this time. Gel extracted using Qiagen protocol with microcentrifuge.
8/11/2011
Heat killed ligase 65°C 20 min.
Discovered that TOP10 cells do not require IPTG for blue/white screen. Spread 25µl 1000x X-Gal/ 175µl H2O onto LB-carb plates, put in 37°C for 1 hour. Followed TOP10 transformation protocol (heat shock), spread one set 50µl cells/150µl LB, one set 200µl cells/100µl LB, put plates in 37°C overnight.
8/12/2011
Blue/white colonies observed for all, except STMutr, which had no growth for both 50µl and 200µl plates. Picked white colonies and restreaked on LB+carb, put in 37°C.
PCR of ß-Carotene producing yeasts to determine which strain is which. The ORFs all have the same promoter and 3'UTR so after PCR with one set of primers I expect to see bands corresponding to each gene.
50µl reaction
| Component | Volume (µl) |
| 2x GoTaq Green Master Mix | 25 |
| Forward primer (10µM) | 5 |
| Reverse primer (10µM) | 5 |
| gDNA template | 200ng |
| dH2O | to 50 |
| Total | 50 |
PCR Program: 2 min @ 95°C, 30x(30s @ 95°C, 30s @ 52°C, 2 min @ 72°C), 5 min @ 72°C
| ORF | Expected Size (bp) |
| crtE | 1131 |
| crtYB | 2234 |
| crtI | 2011 |
| BTS1 | 1008 |

8/13/2011
Inoculated 5ml LB w/streaked out white colonies with remaining promoters/UTRs, put in 37°C drum.
Inoculated 25ml LB w/ TOP10 for competent cell preparation.
8/14/2011
Began preparation of competent cells, but did not have time to finish, so abandoned this.
Miniprepped 8/13 remaining promoter/UTRs.
8/15/2011
BsaI restriction digest of 8/14 minipreps to check if promoters/UTRs ligated correctly.
40µl reaction
| Component | Volume (µl) |
| 10x NEBuffer 4 | 4 |
| miniprep product | 20 |
| BSA (100x) | .75 |
| dH2O | 15.25 |
| Total | 40 |
Incubated @ 50°C 2 hours.

All fragments looked correct, so made 15% glycerol stocks of the E. coli they came from and stored in iGEM -80°C space.
Picked another white colony and inoculated 5ml LB with E. coli containing pTZ19u with STM1utr SmaI ligated in.
8/16/2011
Miniprepped and BsaI digested STM1utr, same protocol as 8/15.
 No fragment observed.
No fragment observed.
8/17/2011
Picked all white colonies (5) from 1st transformation for STM1utr, streaked out on LB+carb, put in 37°C incubator.
The primers for RPS2utr arrived: Did 50µl reaction according to Herculase II protocol with 20µM control primers ZK096/097.
PCR program: 2 min @ 95°C, 30x(20s @ 95°C, 20s @ 58°C, 30s @72°C), 3 min @ 72°C. PCR product to -20°C.
8/20/2011
3/5 white colonies for STM1utr grew, but were too overgrown to pick single colonies, so were restreaked on LB+carb and put in 37°C.
Ran gel of RPS2utr PCR product from 8/16, extracted.

8/22/2011
Inoculated 5 ml LB w/STM1utr (3 colonies), put in 37°C drum.
Gel purified RPS2utr according to Qiagen gel extraction protocol.
Nanodrop of possible RPS2utr product: 8.0 ng/µl.
Loaded 12.5µl (~100ng) in 2% MetaPhor agarose gel to verify product.

Product is too small.
8/23/2011
Streaked out ß-carotene yeasts on YPD, put in 30°C incubator for enzyme assays.
Miniprepped 3 STM1utr samples.
Did overnight BsaI digest @ 50°C according to 8/15 protocol.
8/24/2011
Ran digested STM1utr:

Got one good colony, made 15% glycerol perm of liquid culture (500µl 30% glycerol/500µl cells), put in -80°C w/other promoter/UTR E. coli.
8/25/2011
Another PCR attempt for RPS2utr.
50µl reaction using Herculase II protocol along with 20µM control primers (ZK096/097).
PCR program: 2 min @95°C, (20s @95°C, 20s @45°C, 30s @72°C), 3 min @ 72°C
Ran possible RPS2utr product on gel, extracted.

Gel purified according to Qiagen Gel Extraction protocol, nanodropped cleaned product: 8.9ng/µl. Loaded 11.24µl (100ng) on gel to verify product.

Product looks correct.
8/29/2011
Ligation of RPS2utr PCR product into SmaI-digested pTZ19u.
20µl reaction w/6 molar insert to vector ratio:
| Component | Volume (µl) |
| 10x T4 buffer | 2 |
| SmaI linearized pTZ19u (25.2 ng/µl) | 1 |
| T4 ligase (Enzymatics, 120U/µl) | 1 |
| RPS2utr (8.9ngµl) | .66 |
| dH2O | 15.34 |
| Total | 20 |
Ligation at 25°C overnight.
Streaked out 23 other promoter/3'UTR containing E. coli from -80°C glycerol stocks on LB+Carb, put in 37°C incubator for inoculation of liquid cultures for miniprepping to make stocks of fragments.
8/30/2011
Transformed RPS2utr/pTZ19u ligation into TOP10 cells according to TOP10 transformation protocol. Spread 50µl in 150µl LB and 200µl in 300µl LB on LB+Carb plates w/40µl 40mg/ml X-Gal spread previously in 160µl LB. To 37°C incubator.
9/2/2011
Couldn't come in to take transformation plates out, plates are too overgrown to be any good.
Miniprepped 23 promoters/3'UTRs according to E. Davis protocol.
40µl BsaI digest of minipreps.
| Component | Volume (µl) |
| 10x NEBuffer 4 | 4 |
| miniprep | 18 |
| BsaI (10U/µl) | 1 |
| dH2O | 14.5 |
| Total | 37.5 |
50°C overnight incubation.
9/3/2011
Heat kill BsaI 65°C 20 min. Gel extraction of all parts.
9/9/2011
Gel purification of all parts according to Qiagen gel extraction protocol.
Nanodrop concentrations of products (ng/µl)
| Fragment | concentration (ng/µl) | Fragment | concentration (ng/µl) |
| TDH3p | 14.9 | FCY2p | 22.0 |
| TDH3utr | 6.2 | FCY2utr | 6.0 |
| RPL24Ap | 9.0 | PRY1p | 13.4 |
| RPL24Autr | 4.5 | PRY1utr | 8.1 |
| RPS8Bp | 22.0 | ARD1p | 8.0 |
| RPS8Butr | 5.3 | ARD1utr | 6.5 |
| RPS2p | 18.8 | KRE9p | 6.9 |
| RPS2utr | ---- | KRE9utr | 8.4 |
| RPL8Ap | 11.1 | HHO1p | 7.8 |
| RPL8Autr | 13.6 | HHO1utr | 7.5 |
| STM1p | 15.2 | BAP2p | 15.9 |
| STM1utr | 5.9 | BAP2utr | 19.2 |
Ran 100ng each to verify product.

DNA is not very visible, will do another round of minipreps.
Streaked out another round of 23 promoters and 3'UTRs from -80°C on LB+Carb, put in 37°C.
9/10/2011
Inoculated 5ml LB w/colonies from overnight plates. Plates to cold room, liquid cultures to 37°C drum.
Hybridized loxPsym oligos: Resuspended ssDNAs to 100µM, took 10µl of each in one tube, put @ 94°C 10 minutes, moved to bench and then to cold room for slow cooling.
Nanodropped ss + strand, ss - strand, and ds loxPsym linker. + strand- 1360ng/µl - strand- 1345ng/µl double stranded- 2096.7ng/µl
Ran 1µg of each on 2% MetaPhor agarose gel.

9/11/2011
Miniprepped all 23 overnight cultures according to Marty Taylor's protocol, put liquid cultures in cold room.
9/12/2011
Nanodrop all minipreps from 9/11.
| Fragment | Concentration (ng/µl) | Fragment | Concentration (ng/µl) |
| TDH3p | 292.1 | FCY2p | 116.4 |
| TDH3utr | 288.2 | FCY2utr | 298.4 |
| RPL24Ap | 84.0 | PRY1p | 253.1 |
| RPL24Autr | 385.0 | PRY1utr | 308.3 |
| RPS8Bp | 317.6 | ARD1p | 54.1 |
| RPS8Butr | 235.8 | ARD1utr | 299.2 |
| RPS2p | 338.1 | KRE9p | 290.7 |
| ------ | KRE9utr | 566.7 | |
| RPL8Ap | 307.8 | HHO1p | 292.9 |
| RPL8Autr | 280.5 | HHO1utr | 175.4 |
| STM1p | 27.3 | BAP2p | 262.7 |
| STM1utr | 354.2 | BAP2utr | 337.8 |
Digested all promoters with BsaI, split 3'UTRs into two aliquots, digested one with NotI, then realized that in order to cut the entire 3'UTR and URA3 out I would have to digest with BsaI, which works in buffer 4, while NotI works in NEBuffer 3. Digested the other UTR aliquot with NotI-HF, which works in NEBuffer 4.
9/16/2011
PCR of URA3 w/Herculase. 8 reactions in parallel.
50µl reaction
| Component | Volume (µl) |
| gDNA template (156 ng/µl) | 1 |
| Herculase II polymerase | 1 |
| Herc. II 5x Buffer | 10 |
| URA3 fwd primer (10µM) | 1.25 |
| URA3 rev primer (10µM) | 1.25 |
| dNTP mix (2.5mM ea.) | 5 |
| dH2O | 30.5 |
| Total | 50 |
PCR program: 2min @95°C, 30x(20s @ 95°C, 20s @42°C, 34s @ 72°C), 3 min @ 72°C.
Got 8 very bright bands at correct size (1134bp), extracted all and purified according to Qiagen gel extraction protocol
Nanodropped 8 URA3 gel purification products:
| URA3 fragment | Concentration (ng/µl) |
| 1 | 28.8 |
| 2 | 21.6 |
| 3 | 21.9 |
| 4 | 23.2 |
| 5 | 28.5 |
| 6 | 33.7 |
| 7 | 33.8 |
| 8 | 23.8 |
9/17/2011
NotI-HF digests of all 8 URA3 PCR products.
55µl digest
| Component | Volume (ng/µl) |
| URA3 PCR product | 48 |
| 10x NEBuffer 4 | 5.5 |
| BSA (100x) | .55 |
| NotI-HF (20U/µl) | .3 |
| dH2O | .65 |
| Total | 55 |
2 hour digest @ 37°C, 20 min heat kill @ 65°C.
Ligation of URA3 into 3'UTRs, also a ligation of plain RPS2utr. 20µl ligation (same protocol as 7/30. 1 hour at 25°C. Transformed into DH5α competent cells (protocol). Spread two volumes of cells, 50µl cells in 250µl LB and 200µl cells in 300µl LB.
9/18/2011
Transformations showed mixed results. Many plates showed only white colonies. Picked 7 colonies from good plate and 2 colonies from bad plates for miniprep and digestion to check for URA3 ligation. Inoculated in 5ml LB+carb.
9/19/2011
Miniprepped 9/18 liquid cultures [[1]].
9/20/2011
BsaI digests of URA3 3'UTR ligations.
50µl digests
| Component | Volume (µl) |
| Miniprep DNA (~1.2µg/µl) | 1 |
| 10x NEBuffer 4 | 5 |
| BSA (100x) | .5 |
| BsaI (10U/µl) | 1 |
| dH2O | 42.5 |
| Total | 50 |
9/23/2011
Ran gels of 9/20 digests.








Made SC-his-leu-ura plates for Vitamin C transformations.
| Component | Volume (ml) |
| 2x SC--8 | 250 |
| 20% galactose | 50 |
| 40mM trp | 5 |
| 16mM ade | 5 |
| 100mM lys | 5 |
| 50mM met | 6 |
| 4% agar | 250 |
9/30/2011
Finally had time to look at the 9/23 gels. Turns out there is an internal BsaI site in the URA3 sequence, so will have to find a BsaI/BsmBI free URA3 sequence and redo the 3'UTR tagging.
10/1/2011
Ligation of RPS2utr into pTZ19u.
10/2/2011
Picked 5 colonies from each transformation (3), and inoculated 5ml LB+carb.
10/3/2011
Made 15% glycerol stocks of the possible RPS2utr bacteria. (No time to miniprep)
10/7/2011
Restreaked the 15 possible RPS2utr strains onto LB plates.
10/8/2011
Inoculated 5ml LB+carb with RPS2utr candidates.
10/9/2011
Didn't have time to miniprep today, will redo next week.
10/16/2011
Miniprepped the RPS2utr candidates, put into -20°C.
10/23/2011
Digested yeast shuttle vectors with 4 igem sites to verify single cuts.
10/28/2011
Inoculated VitC constructs with Rob to miniprep and make expression cassettes.
Violacein Biosynthesis in Yeast
June 7th, 2011
Purpose:
- Make 4 copies of violacein building blocks and select promoters and 3’UTRs on 96 well plates. 2 were made previously by Dr. Boeke's Lab Tech and I (not entered into wiki). Therefore I am only making 2 new copies today.
- Begin growing colonies on petri dishes (to prepare for miniprep).
Procedure:
- Made 100ml LB+Carbenicillin Solution (ampicillin resistance gene also resists Carbenicillin) using 100uL 1000x Carbenicillin (thawed from -20 degrees Celsius) and 100mL LB Broth.
- Labelled plates and Petri Dishes
- Except for one petri dish (forgot to add a label), each label says: “iGEM: Boeke promoter, 3’ end & Vio BB’s from YW (pseudonym); F2.”
- Transferred LB+Carb solution to a ‘sterile reagent reservoir for multi-channel pipetting.
- Took out a previous copy of the genes (arrayed on a 96 well plate) from -80 degree freezer, placed in a centrifuge with a balance.
- Centrifuged until speed reached 1000g (short spin).
- Used a 12-lane multichannel micropipette to transfer 100uL of LB+Carb to rows A-D on new 96 well plates.
- Placed air-pore tape on each new 96 well plate (now inoculated) and incubated them at 37 degrees Celsius.
- Incubated petri dishes at 37 degrees.
Problems:
- I added LB+carb to every well in rows a-d. The original array only filled row A/C, half of row B and 11/12 wells in Row D.
- Always remember how original array was organized and always stick to it.
- Original plate (the one being copied) was left out for around 50min. Not good.
- Next time, only take out plate from -80 after each new 96 well plate/petri dish has been prepared for inoculation (e.g. LB+Carb should be added to wells before original plate is taken out to thaw).
- I am still learning where everything in the lab is. This will take time.
- When pipetting, I would scrape the tip onto the side of the well to get rid of the final drops. This motion resulted in me almost mixing the original cultures.
- Remember: Do not worry about “pipetting the last drop.”
- When inoculating petri dishes, the liquid sprayed out of the tip instead of dripping out.
- Don’t let the liquid spray out of the tip, the different cultures may mix.
June 9th, 2011
Purpose:
- Inoculated test tubes (filled with ~ 5mL LB+Carb)with each part (Vio ORF Building Blocks (ie ORF fragments), Promoters and UTR's)
Procedure:
- For each part, took wooden stick, placed flat end onto a single colony on the agar plate, dipped stick into the appropriate text tube (inoculating the test tube) and threw out the stick.
- Incubated them overnight at 37 degrees on a shaker (a slanted spinning wheel with slots for test tubes).
June 10th, 2011
Purpose:
- Mini-Prep of parts (Vio ORF Building Blocks, Promoters, UTR's)
- Double Cut Restriction Digest of Mini Prep Samples (to run an analytical gel to check if the correct inserts (parts) are present).
Procedure:
- See File:Miniprep stet prep pm .pdf for boiling mini prep.
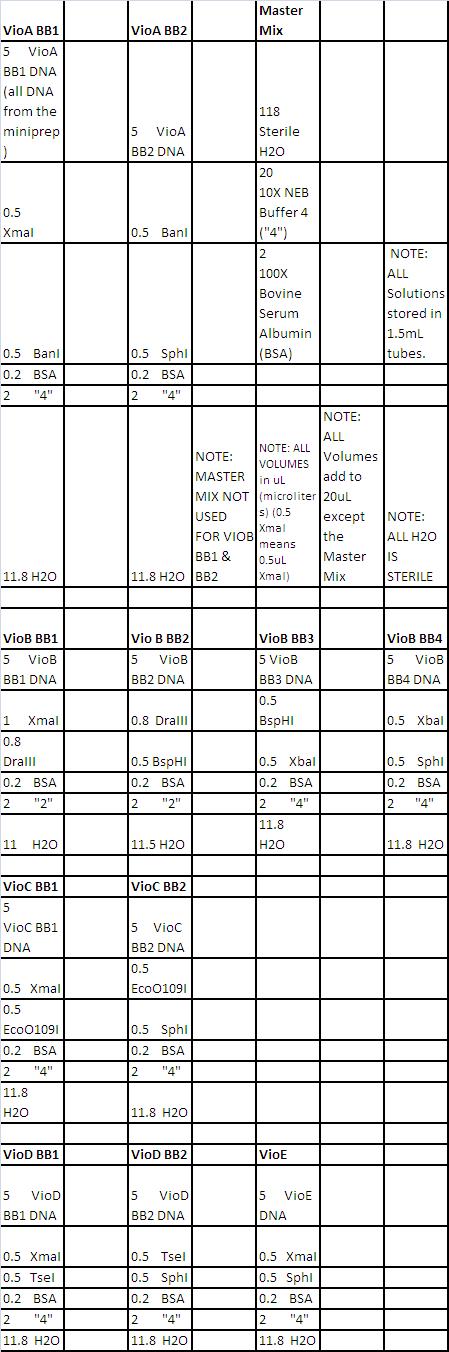
- See Double Digest Reaction Mixes here
- All other parts (promoters and UTR's) double digested with XmaI and SphI)
- See neb.com for correct digestion procedures with each enzyme.
- After digestions completed, stored samples at -20 degrees Celsius.
{kind=link}
Results:

Results show high concentrations of plasmid, too high. In reality there is RNA in the solution too and nanodrop detects nucleic acid without distinction for DNA or RNA. According to Dr. Boeke’s Lab Tech, the readings still indicate that the DNA concentration is probably high.
Problems:
- Mini Prep:
- Instead of boiling all 29 samples at once, I boiled 15 and then 14. For the first 15, I boiled for 2min 45sec and not the required 3 minutes. The reason: the water was not boiling when we entered the samples. At the last minute, we had to refill the bath with hot water and place the samples in. We set the timer for 4 minutes and the water started to boil with 2minutes 45seconds left.
- Sometimes during mini prep, I did not change the tip for each sample while aspirating supernatant.
June 11th, 2011
Purpose:
- Gel Electrophoresis of Yesterday's Digests
Procedure:
- Added 4uL CS Red Dye (with added RNAse) to each sample and 2 log ladder.
Problems:
- Added 7uL Dye to VioA2
- Ladder Didn't show up on the gel (must redo tomorrow).
June 13th, 2011
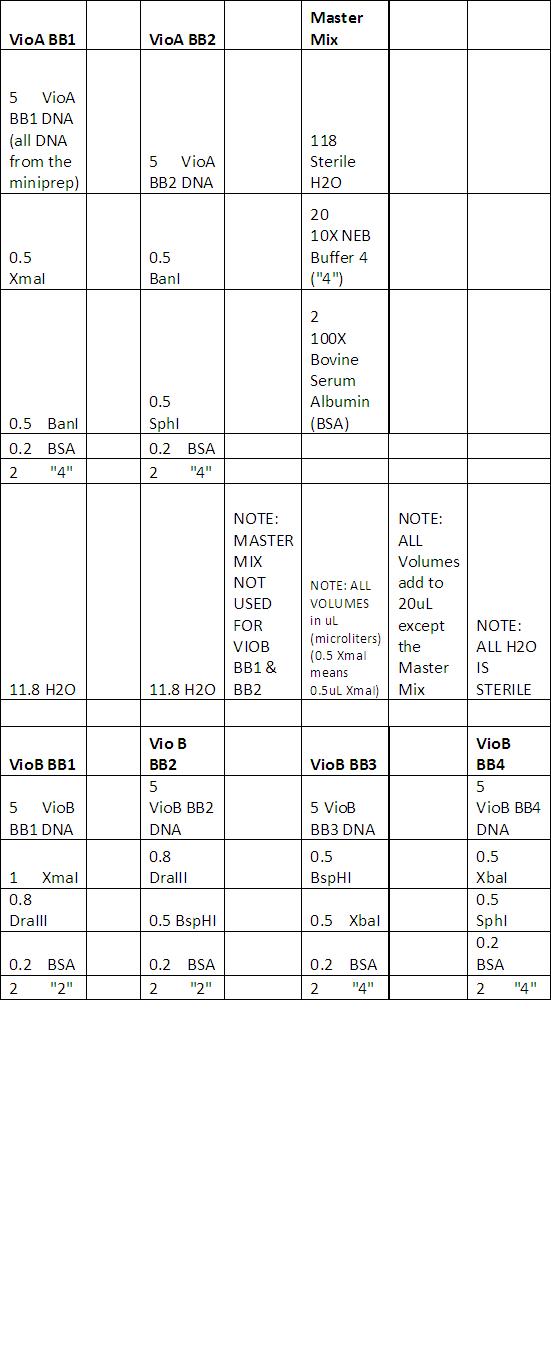
Purpose:
- Redo Digest (No image uploaded), the ladders on the gel did not show up. Will use 2 log ladder with blue Juice this time and not CS Red. Will add a second digest condition for VioE, using a BsaI single cut.
- Note: Although the ladder couldn't be seen properly, we agreed that the patterns/digests were correct and matched what we expected. We believe there is nothing wrong with our restriction enzymes or our starting material (parts).
Procedure:
- See reaction here
- See neb.com for correct digestion procedures.
{kind=link}
Problems:
- Accidentally heated VioE2 tube with the others at 37 degrees. After 37 degree heating, I Placed it in 50 degree water bath for around 1hour 55minutes. Made a PCR tube of new VioE2 solution (according to above table) and used thermocycler (PCR machine) to heat it for 2hours at 65 degrees and refrigerate it (4 degrees) afterwards). Still not the right temperature. I made yet another PCR tube of VioE2 (VioE2=VioE cut with BsaI) and 'PCR'd' at 50 degrees (I refer to this tube now as 'VioE BsaI cut (3).'
- "R" from Zhu lab took VioD BB’s and VioE2 out for me and placed the samples in my minus 20 freezer space. I left the lab when I put them to digest.
June 14th, 2011
Purpose:
- Run a preparative gel (a gel for gel extraction/purification of Vio ORF Building Blocks).
Procedure:
- Made 200uL of 1% agarose gel with 1x Ethidium Bromide (diluted from 20000x) added.
- 25 thick lanes indented into gel.
- Planned Lanes:
![]()
- Note: Lane 25 for this run was for VioE BsaI Cut (3) (see June 13th's 'Problems' section).
Results:
- Gel looked fine. Got expected band patterns. See todays 'Problems' section
Problems:

- It's not good practice to take a picture of a gel before gel extraction, UV exposure can damage the DNA. Hand held UV light can go to long wave, unlike the 'picture taker, which uses only short wave UV light' Long form is less harmful to the DNA. This gel must be re-done.
- Note: New practice is to allow pictures before gel extraction, but exposure to short wave UV light must be as short as possible (less than a split second is good).
- See 'Blooper' section. As we exposed the gel (today's image on the wiki) for a short time to short UV, we could have used it for gel extraction. Unfortunately, it fell and shattered.
- I redid the digests according to the June 13th protocol to run a future analytic gel (to definitively see if the patterns are what we expect).
- Note: I also ran another exact set of digests in parallel for a future preparative gel for gel extraction.
June 15th, 2011
Purpose:
- Run an analytical gel to determine if the digests worked before we do a preparative gel.
Procedure:
- Made 2x 200mL 1% gels with EB, exactly like the June 14th Gel.
- Made a 100mL gel with 25 thick, narrow lanes. We decided that we should use a smaller gel for an analytical gel. This way (out of 1 tube of 25uL digest mixture + CS Red) we can load 3uL for the analytical gel and the rest for the preparative gel. I originally made a copy of each digest; one for analytical, one for preparative. We decided to use one copy for both (the second set of digests I made remain unused).
- 2 log ladder in lane 2, 100bp ladder in lane 24.
Data:

Problem:
- Accidentally switched the 2nd copy Vio B BB1 (which should have remained unused) with the first copy of VioB BB2 (which should have been loaded). It was a mis-labelling error. There are 2 vio B BB1 samples in the analytical gel (lanes 5 and 7), and no VioB BB2. We decided we could proceed to the preparative gel step as I found the 1st copy VioB BB2.
June 19th, 2011
Purpose:
- Plan with Zheng how to do a ligation reaction (I planned today to digest the vector in preparation for the gel extraction and future ligation of ORF buidling blocks).
- Final Planned Procedure for Ligation:

- Note: If using enzymatics reagents, ligate at room temp for an hour. If NEB reagents, 16 degrees Celsius (from now on, all temperature measurements are in Celsius) overnight.
June 20th, 2011
Purpose: Gel Extraction
Procedure: See Qiagen Gel Purification protocol, June 13th Gel Specifics
- Only 16uL VioE 2x RE Cut loaded into the gel.
- 120V 15min, 10V 15min, 130V 10 Min (all to try and separate 2 bands, only 1 of which we wanted to extract.
- Left for home after QG, NaAcetate and 100% Isopropanol were added to the excised gel fragments (experiment not finished yet). A post-doc said this is OK to do.
June 21st, 2011
Purpose:
- Finish gel purification
Results:
- DNA concentrations are small, but workable. I digested 30uL of each ORF with 1uL of each enzyme and added dye just in case.
June 22nd, 2011
Purpose:
- Just came in to add dye to the VioD digests (they need to be digested at 2 temps, which is why I didn't add dye to them with the others).
June 30th, 2011
Purpose:
- Replica Plate transformed colonies from LB+Carb plates to LB+Carb+XGal+IPTG Plates.
Procedure:
- Mixed 3ml LB with 500uL 1000x IPTG and 500uL 1000x XGal.
- Spread 19X200uL aliquots of this solution onto 19 LB+Carb Plates using sterile glass beads.
- Note: I selected 15 out of 31 plates for replica plating and made 4 extra Xgal plates just in case.
- Dr. Boeke said to let transformed colonies grow more at 37 degrees.
- Left the 15 selected plates in the 37 degree incubator, left the new xgal plates to soak on my bench, placed all other plates in the 4 degree fridge.
Problems:
- Wasted lots of time thawing the xgal because its solvent is DMSO and I left it on ice to thaw. Note: Never leave DMSO on ice to thaw, it will not melt. Do not even leave thawed DMSO on ice, it will refreeze quickly.
- Be more careful when pouring beads, wasted a couple.
July 1st, 2011
Purpose:
- Replica Plate transformed colonies from LB+Carb plates to LB+Carb+XGal+IPTG Plates (made yesterday).
- Inoculate glass tubes for mini prep for each white colony on the xgal destination plates.
Procedure:
- Drew a line on original plate and destination plate. Used it to make sure destination plate colonies had the same orientation as original plate. Looked for white colonies in the evening.
- Pressed original plate onto a sterile velvet, pressed empty destination plate onto the newly 'dirty' velvet to pick up the colonies.
- Filled glass tubes with 5mL LB+5uL Carb, labeled them according to ligation condition (16 degree or room temp incubation), transformation volume (e.g. 180uL transformation mixture added to the plate) & sample name (e.g. VioA, VioB,...VioE).
- For all white colonies on the destination plates, I found the colony location on the destination plate and picked it with a wooden stick(pressed the flat end onto the colony, avoiding small satellite colonies). I then shook the picked colony in the LB mixture vigorously, threw the toothpick, and placed the tubes in a 37 degree shaking incubator overnight.
July 2nd, 2011
Purpose:
- Miniprep from yesterday's inoculated tubes.
- Digest
Procedure:
July 3rd, 2011
Purpose:
- Run an analytical get to see if the correct inserts are present.
Procedure:
- Prepare a 200mL 1% agarose gel with EB and 25 think lanes.
- Add 10uL of digest and CS red dye.
- Run at 150V and take picture after 40min.
Results:

- C, D, E ligated well, but E-90-R did not ligate.
- Dr. Boeke suggests making replica plates of all other A, B plates and searching for a white colony.
July 5th, 2011
Purpose:
- Make 14 replica plates of A/B
Procedure:
- Used 25mL of agar, 25uL of IPTG and Xgal, and 150mL LB
- Mix consists of 375uL of Xgal, 375uL of IPTG, and 2250uL of LB for a total volume of 3000uL.
- 30, 90, and R 30uL plates held at 16 degrees Celsius.
Results:
- VioA and VioE 30uL RT had no colonies.
- Because the original colonies were very small, the 90uL B was not re-plated to allow it to grow more.
- All colonies incubated at 30 degrees Celsius to try to lower the amount of Halos.
- Note: Halos: Original colony secretes resistance protein for other colonies to leech off of. Only good colonies survive in liquid suspension, as Halos can't effectively leech anymore. Streaking on a new plate kills most Halos too.
July 6th, 2011
Purpose:
- Find whites on replica plate B-90 1b and inoculate the white on the A XGal plate
- June 27th ligation: aliquot 5uL and save, leave the rest overnight.
- Transformation (June 27th protocol)
- Protocol:
- Transform dilute PTE 19 after first A/B tubes heat shocked.
- Put ligation tubes back to 16 degrees Celsius after heat shock of vector and negative control.
Results:
- One plate of A-30-RT has a white-ish colony (Placed in 37 degrees Celsius to see if it turns blue
- All useless inoculated plates from yesterday thrown out (except the whitish A, which was put in 4 degrees Celsius).
- No DNA control full of bacteria
- PTE19 plate empty
- PTE-LB plate chalk full
- Conclusion: No DNA plate and PTE19 plate (LB-Carb) switched. No DNA bacteria went to PTE19 plate and vice-versa. PTE cannot be toxic as it grows on LB.
July 7th, 2011
- Protocol:
- Inoculate from July 5th VioB (85uL, 45uL at 16 degrees Celsius) and July 30th VioA (180uL at 16 degrees Celsius)
- Replica plate July 6th VioA (30-90-180 at 16 degrees Celsius)
- PTE19 plate in 4 degrees Celsius and negative DNA and PTE LB (proof)
Results:
- "All whitish A" from yesterday was blue...not using it.
- July 5th 15uL VioB had ambiguous large colonies, want to let it grow
July 8th, 2011
Purpose:
- Miniprep B-T 16-285 a, B-T 16-245 & A 16 180 (Take 3mL of each and split into 2 1.5mL tubes)
Procedure:
- Aided STET suspension by heating at 37 degrees Celsius for 5min
- After double digest, run 100mL 1% agarose gel with EB for 25min
- Inoculate relica whites if gel doesn't work
- Vortexed B-T456 and A minipreps before adding to digest mix
- Run 50mL 1% agarose gel with EB for 25min at 150V
- Lanes: Ladder, A, B-45, B-85, Ladder
Results:

- Inoculated B-T 16-285 b and c were clear, with no bacteria
- 15uL VioB had no colonies
- VioA 185 and B 95 had one white colony each
- Gel broke, but probably still worked
- Dr. Boeke suggests digesting with the middle restriction enzyme (for the unique restriction site that stitched the BBs together)to make sure to make competent cells, design primers to add BSAI sites and unique sequences for each Vio expression cassette objective, and stitch all cassettes to the neochromosome in one go.
- Suggestions: Whenever measuring pieces, always digest the vector alone and run it alongside the others. Also, run gels longer, add EB to TE.
July 9th, 2011
Purpose:
- Make sure no internal BsaI or BsnbI sites are in any designs.
- Double digest within regions (no E required, at least 3 Bs)
- Single digest with BsaI
- Choose the more plentiful Eu out of XmaI and SphI
- Design primers according to the incoming Boeke diagram
July 10th, 2011
Procedure:
- Run gel at 150V for 1 hour (500mL, 1% agarose)
- Lanes: Ladder, A B C D (BsaI), A B B2 B3 C D, Ladder
- Add 400mL TE and and EB
Results:

- Digest went too long, need to redo
- BsaI and DraII at 37 degrees Celsius for 50min
- XbaI is gone - finished, will stick with a DraIII, BspHI 28cut
- Make mixes
July 11th, 2011
Purpose:
- Make 500uL of CS red dye with 0.1ug/uL of RNAse
- Digests
July 12th, 2011
Purpose:
- Run gel at 150V for 75min (1% agarose with EB in gel and buffer, 5uL CS red dye per sample)
- design Boeke oligos
- Protocol:
- Lanes: Ladder, BsaI A B B B, Ladder, BsaI C C D D, Ladder
Results:

July 13th, 2011
Purpose:
- Run 50mL 1% agarose gel with EB (300mL TTE & 15uL EB for 45min at 150V)
Results:
- VioB is not here :( Transform overnight ligations, miniprep and try again
July 19th, 2011
Purpose:
- Digest and ligate B 1, 2, 3 to PTZ19u tomorrow
- Transform overnight ligations
- Design oligos
July 23th, 2011
Purpose:
- Ligations
- Finish oligos with Boeke
Procedure:
- Cooked vector at 65 degrees Celsius for 20min
Results:
- Will aliquot 6uL for transformations. Leave rest overnight
- Make sure melting temps for yeast DNA regions are nearly equal +/- 1-2 degrees best (so that lengths are nearly equal)
July 26th, 2011
Purpose:
- Cook all my ligation rxns for 10min at 65 degrees Celsius
- Digest available ORFs with BsaI in preparation for gel purification
- Strategy: Take bare minimum (may need to redo)
July 27th, 2011
Purpose:
- Gel purify ORFs
- Digest PTZ14u with BsaI to load with other 4 samples
Procedure:
- Gel lanes: Ladder, A C D E PTZ19u
Results:

- A is suspect, may be contaminated with upper band
- C is inseparable on this gel
- Mixed D and E, must redo and repeat July 26 digest
- Run bigger and longer gel next time
July 28th, 2011
Purpose:
- Gel extractions on long plate (300uL TTE and 3g agarose and 15uL EB)
- Procudure:
- Run for 1.5hr at 150V
- Lanes: Ladder, A C, Ladder, D E, Ladder
- Make new wide, thin gel (1.5% to run at 100-120V). Tape combs together and add 12.5uL per well.
Results:
- Blue band at ~300bp, gel was slow
July 29th, 2011
Purpose:
- Run gel for 2hrs
- Purify what you can. If time permits, set up a double digest.
Results:

- DNA did not diffuse from the refrigeration. (Don't try to do too much in a day)
- A and D melting gel at 48 degrees Celsius, closest block available
- A: 3.2ug/uL
- D: 9.1ug/uL
- C: highly suspect of contamination, 10.8ug/uL
August 1st, 2011
Purpose:
- Transform B1, 2, 3
- Make 6x XGal plates (1 for 2hrs, one for overnight)
Procedure:
- 2hr: 50 and 500 uL
- Overnight: 50 and 500uL (no DNA, just vector only)
August 2nd, 2011
Purpose:
- Transform B 1, 2, 3
- Digest promoters with BsaI
- ligate to appropriate ORFs
August 4th, 2011
Purpose:
- Digest vector with XbaI and XmaI at 37 degrees Celsius for 0.5hr.
Procedure:


- Cook digest, run 50mL 1% agarose gel with EB of all BBs (ligation mix, individual parts, ladder, and uncut vector (all>10ug))
- Transform with 1ug uncut vector
- Larger than 30uL...insert digest
- After 2hrs, check on gel and leave on for 3hrs.
Results:
- some wells too shallow
August 5th, 2011
Purpose:
- Ligation and transformation
Results:
- Did not digest vector first
- added wrongly-digest vector
August 6th, 2011
Purpose:
- Prep PTZ19u for ligation
- Prep ligations to add digest vector
Procedure:
- 200uL PB, vortex, column, spin for 1min, discard, add 750uL of TE, spin for 1min, discard, spin for 1min @1300rpm, discard, add 25uL EB, spin 1min (into new tube), nanodrop
- Add 25uL of water, 3uL T4, 3uL ligase. Split to three tubes, each containing 20uL. (Label 1:3, 1:5, 1:10)
- Add 1, 2, 3 to right tubes, add 1 to vector tubes.
- Ligate 2hrs overnight, transform 1uL, ligate
August 9th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- Ran a gel to verify size/presence of VioB Building Blocks in previous gel purifications.
- It’s not on my notes, but according to the brightness of the bands, I think I ran 1ul for the Elu product and 3 ul for B1 B2 B3 B4
* ladder B1Elu B1 B2Elu B2 B3Elu B3 B4Elu B4

- Redo Ligation: Purpose is to get VioB by ligation
- Rxn1: B1:3ul B2:3ul B3:3ul vector:1ul T4 ligase:1ul T4 10xBuffer:2ul H2O:7ul Total=20ul
- Rxn1Control1: B1:3ul B2:3ul B3:3ul XmaI XbaI vector:1ul T4 10xBuffer:2ul H2O:8ul Total=20ul
- Rxn1Control2: XmaI XbaI vector:1ul T4 ligase:1ul T4 10xBuffer:2ul H2O:16ul Total=20ul
- Rxn2: B1:3ul B2:3ul B3:3ul B4:3ul vector:1ul T4 ligase:1ul T4 10xBuffer:2ul H2O:4ul * Total=20ul
- Rxn2Control1: B1:3ul B2:3ul B3:3ul B4:3ul XmaI SphI vector:1ul T4 10xBuffer:2ul H2O:5ul * Total=20ul
- Rxn2Control2: XmaI SphI vector:1ul T4 ligase:1ul T4 10xBuffer:2ul H2O:16ul Total=20ul
Ligate for 2 hours
- Then use LA Taq to do PCR.
cocktail and DNA:
- Ligation DNA 1ul
- PCR Rxn Mix
Primer1: 1ul Primer2:1ul La Taq: 0.5ul 10x Buffer: 5ul dNTP: 8ul H2O:33.5ul Total: 50 ul
- PCR condition:
94 degree 1 min 30 cycles: 94 degree 30 seconds 55 degree 30 seconds 72 degree 1 min 72 degree 10 min 4 degree infinitely
- PCR Gel:

- It’s hard to tell if there’s any band below the first well. I inverted, and adjusted many times on the picture taking machine and saw a super light band which can’t be shown on the picture.
- It’s not on my notes, but I sense this is a picture of gel which contain 2 product, the new VioB is the PCR version of my ligation product, the old VioB may be yours ligation product which I might also PCRed.
- In the following picture, only 2 wells are for iGEM. Those 2 wells proved that puc19 vectors were purified. Other wells are for my personal projects in the Boeke Lab.

- Lastly, transformation: 4 controls and 2 rxns and 1 no DNA control
August 10th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- “We’re waiting for the transformation from 2 a.m. Transformation efficiency isn’t very high. Rxn 2 with B1 B2 B3 and B4 only has one colony. We did some troubleshooting with Qing (Member of the [http://www.bs.jhmi.edu/MBG/boekelab/members.html Boeke Lab]). She said that our vector band is really bright, so its concentration is probably very high and our pieces of VioB [are not very] concentrated. Although we used 3:1 volume ratio of B1 B2 B3 B4 and the vectors, it’s not good enough.”
- Plates Images: (probably saw colonies around 4 p. I think I incubated them at 37deg.)

August 11th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- Addition to August 10th—picked a white colony for liquid culture in the 37 degree shaker. I then used Patrick Cai’s Miniprep protocol and stopped at the first step.
- I transformed 5ul ligation product by using Patrick’s competent cell which are super hot as Qing suggested. And put them into 37 deg overnight.
- And we used herculase enzyme and protocol to do another PCR of VioB. We got a super bright band:

- The upper one is around 3kb. There is a double band situation. May be need further explanation.
- Then I transformed this PCR version VioB into E.coli, I believe it’s Patrick’s competent cell. He tested them they’re super hot. Make them overnight at 37 degree. We get them next morning I believe.
- Here’s the plates:

August 12th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- I continued miniprep of VioB’s first time transformation on 8/9. (The time with only one white colony of B1 B2 B3 B4 + vector)
- Then I digested the minipreped white colonies from first time transformation of VioB.
I checked with the [http://biologylabs.utah.edu/jorgensen/wayned/ape/ ApE(Universal)computer program] of how to digest.
- Digest Rxn Mix:
1 x Rxn 10 x Rxn
DNA 2 ul / EcoRI 0.25 ul 2.5 ul Buffer 4 2 ul 20 ul 10 x BSA 2 ul 20 ul H2O 13.75 ul 137.5 ul
- Aliquot 18 ul per tube. Incubate @ 37 degree for an hour.
- Run gels of the miniprep: (Rxn11 means colony 1 which has been circled and numbered on the plate of Rxn 1, Rxn12 13 14 15 mean the same thing; R1C2dot?(Rxn1 Control2 dot question mark))
- Gel lane order:
ladder Rxn11 Rxn12 Rxn13 Rxn14 Rxn15 Rxn21 R1C2dot? R2C1dot?1 R2C1dot?2
- Gel:

- The plates I made on 8/11, the ones with 5 ul ligation product transformed. I believe what I used on 8/9 is 1ul ligation and 200 ul competent cells. This time is five times volume of ligation product than the first time transformation. It’s the same ligation from 8/9.
- Plate Pictures:

- I dip some colonies into a growth plate. 100ul LB/Carb in sterile 96 well plates. Overnight at 37 degree.
* Plate Schematic:

- I picked 24 colonies from VioB PCR 2nd time, 24 colonies from VioB Rxn1 2nd time, 12 colonies from VioB Rxn 2 2nd time.
- I did overhang PCR of ACD, because primers for E didn’t arrive with them. I diluted primers which are in Patrick’s primer box. There’s a concentration number on the commercial tube, for example, 25.1 nM, multiply by 20, 502ul is the volume of the water I added to the commercial tube. Then I made another stock to eppendorf tubes which concentration I can’t remember. Ask Patrick for them.
- Checked the size of ACD
A: 1295+500 C: 1328+500 D: 1160+500
- Used herculase and protocol:
- 50uL rxn mix:
1uL Exp. Cassette DNA 1.25uL forward primer 1.25uL reverse primer 10uL 5x Herculase buffer 0.5uL Herculase 4uL dNTP 35.5uL H2O
- Rxn:
94 degrees-2min
30X Cycle 94 degrees-20sec 55 degrees-20sec 72 degrees-20sec
72 degrees-3min 4 degrees-Infinite
- After PCR:
- We can tell the only white colony worked, it’s 3kb. (Rxn2)
August 13th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- Gel purified VioA C D
Weight: A:0.35g C:0.37g D:0.42g
August 15th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- I wanted to verify the VioB I got so that I can be more sure. Digested internal restriction sites in the VioB ORF Sequence.
- Double Digest Reaction Mix:
DNA 2ul Enzyme 0.25+0.25 ul Buffer 2 ul 10xBSA 2 ul H2O 13.50 ul
- Only one exception, when I digested with BspHI and XbaI:
DNA 5ul BspHI 0.5ul XbaI 0.5ul NEB Buffer 4 2ul BSA 2ul Sterile Water 10ul
- I did this first then I changed to the condition above for other enzymes.
- BspHI XbaI Digest Gel:

- The other gel:
* order:
ladder XmaI BspHI XmaI XbaI XbaI SphI BspHI SphI
VioB internal site digestion Gel:

- They are not perfect match, but Patrick said they’re fine.
- Then I dilute primers for VioE to do overhang PCR.
24.3x20=486 22.1x20=442 then get 10 ul our of each of them, add 40 ul water to make 10uM stock.
- Overhang PCR:
Dilute Template PCR 1:20 (1ul VioE+19ul H2O) cocktail:
5x Herculase Buffer 10ul Herculase Enzyme 0.5ul dNTPs 4ul H2O 35.5ul
Diluted DNA 1ul primer1 1.25ul Primer2 1.25ul cocktail 47 ul total 50ul
- PCR condition:
94 degrees-2min
30X Cycle 94 degrees-20sec 55 degrees-20sec 72 degrees-20sec
72 degrees-3min 4 degrees-Infinite
- Then ran the gel, pretty ladder. VioE may be contaminated so didn’t show anything:

- I shake VioB PCR and ligation-transformation product from growth plate to this big tube I left you. Because Dr. Boeke advised that I not do csPCR with them. They were shaking in 37 degree overnight.
August 16th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- Tubes are ready for Miniprep. But I only did 6 for miniprep, they’re from VioB Rxn2 2nd time plate which contains B1 B2 B3 & B4.
- And there’s a correction for the previous day's entry, I said there are 12 colonies I dip from VioB Rxn2 2nd time plate, but actually there were only 6.
- I dip some colonies into a growth plate. 100ul LB/Carb in sterile 96 well plates. Overnight at 37degree.I dip some colonies into a growth plate. 100ul LB/Carb in sterile 96 well plates. Overnight at 37degree.
- 96 Well Schematic:

- I picked 24 colonies from VioB PCR 2nd time, 24 colonies from VioB Rxn1 2nd time, 6 colonies from VioB Rxn 2 2nd time.
- I ran a gel for VioA C and D which already purified:
* Order:ladder A C D, 3 ul DNA+3 ul loading dye

August 17th, 2011
Experiments Performed by Xin Yuan (see Acknowledgments ). These entries are transcribed from her lab notebook.
- I nanodropped all the miniprep product, the concentrations are on the tubes. And ran a gel of them:

- Forgot to digest before running the gel and order is ladder VioB no PCR 1 2 3 4 5 6 Qing’s stuff ladder. Lane 3 is wired.
August 22nd, 2011
Purpose:
- Draw up offcut ligation for RFC assembly standard
Procedure:
- Digest 30uL E180 16-1 (7/2/2011) and run it by two purified ORFs
- Run 20-well gel (thin comb, B2-20, 200mL 1% agarose with EB). Load 50ug
Results:
- Purify E, use rest of digest product
August 23rd, 2011
Purpose:
- End Goal: 58ng scale ligation of VioE to its TDM3p and RPL8a utr
Procedure:
- Gel purify what's left of digest (took 10uL out of 40uL solution)
- Run 7x8cm gel (100uL, 1% agarose, and 5uL EB)
- Digest 10uL of each promoter/utr
Results:
- Prep Gel Pic:

August 24th, 2011
Purpose:
- Overhang PCR
August 25th, 2011
Purpose:
- Analytical gel of VioE cassette PCR product
Results:
- Gel Pic:

August 26th, 2011
Purpose:
- End Goal: Ligate VioB ORF to RPS2p & utr (5ug for each part)
- Gel purify ORF after BsaI digest
- Digest p & utr
Results:
- Used wrong tube
- Gel Pic:

August 29th, 2011
Purpose:
- Digest VioB DNA with BsaI and BsaI/XbaI
Procedure:
- Run 7x8 1% agarose gel for 35min at 150V
Results:
- Pre-Gel Extraction:

- Post-Gel Extraction:

August 30th, 2011
Purpose:
- Overhang PCR of VioB expression cassette
Procedure:
- Recommended extension time is 2min at 72 degree Celsius
- Run 6-well 50mL 7x8 1% agarose gel at 150V (add 2.5uL EB)
- Lanes: Ladder, 0.5 1 2, Ladder
Results:
- Bands turned out correct
- Gel Pic:

September 1st, 2011
Purpose:
- End Goal: Design, Digest, and Ligation of all 5 Vio genes
- Nanodrop all products
Procedure:
- Ligated according to Aug 23rd protocol. Left at room temp.
Results:
- Lost VioE, re-PCR and gel.
September 2nd, 2011
Purpose:
- PCR cleanup (Invitrogen)
- Design ligation, find PUC19 to digest and do it
- Get reagents for yeast transformation
- Grow culture (inoculate 10mL YPD with 2uL cells for tomorrow, measure OD and dilute based on murkiness on Sunday)
Procedure:
- Run 7x8 50mL 1% agarose gel + 2.5uL EB at 150V for 35min
- Dilute YPD culture, multiply dilution factors by the absorbance reading
Results:
- Gel Pic:

September 6th, 2011
Purpose:
- Final Plan: Select Auxotroph specific vector, get sequence
- Re-PCR expression cassettes and digest internal sites
September 7th, 2011
Purpose:
- Get PRS416 DNA, digest with EcoRI and HindIII
- Find smallest possible bar and comb
Procedure:
- Run 150mL 1% agarose gel with 7.5uL EB at 160V
- Lanes: 300ug 200ug, Ladder, 100ug 50ug 25ug 10ug 5ug 1ug
- Lanes: 300 200 100(1) 100(10) 50 25 10 5 1(1) 1(10)
- Lanes: 10 50 300 200 100
Results
- Water makes ladder float, not sink
September 8th, 2011
Purpose:
- PCR cleanup products (amplify them)
- Gel purify PRS416
- PCR Purify again
- Gel purify
Procedure:
- Run 7x8cm gel(50mL, 1% agarose, 2.5uL EB)
- Lanes: Ladder, A B C D E
Results:
- Gel Prep Pic:

- Analytical Gel Pic:

- A, B, C, D good, E is too small (don't purify)
- Redo E, likely primers were wrong
- Use 1uL PRS as positive control in transformation
- Redo PCR (cleanup took out final product)
September 9th, 2011
Purpose:
- Transform ligation misses and no DNA or vector control
- Redo PCR for VioE
Procedure:
- Add 5uL positive and negative ligation and 1uL PRS416
- 2min on ice
- Heat shock for 45s
- Ice for 2min
- Added 450mL SOC to each
- 1hr on shaking incubator
- 1 plate (50uL), next plate (centrifuge 7000rpm for 25s)
- Take 350mL supernatent out, plate rest after resuspension with pipet
Results:
- Can't tell which tube was PRS416, and no DNA.
September 10th, 2011
Purpose:
- Miniprep colonies and digest them
Procedure:
- Run 7x8 50mL 1% agarose gel with 2.5uL EB
- Lanes: 5uL Ladder, 10uL each of E(new) and E(old)
- Redid VioE PCR from ligation mix
- PCR cleanup with Qiagen
- Gel purify of undigested PCR clean product
- Run 12x14 150mL 1% agarose gel with 7.5 EB
- Lanes: A, Ladder, B X, Ladder, C X D, Ladder, E X X
Results:
- Gel Pics:


- Amplified the wrong fragment?
September 12th, 2011
Purpose:
- Colony PCR design
September 13th, 2011
Purpose:
- Colony PCR
Results:
- Gel Pics:


September 15th, 2011
Purpose:
- Religate in case yeast transformation didn't work
- Colony PCR
September 16th, 2011
Purpose:
- Colony PCR gel (each 96-well plate section of 3 rows occupies one gel row)
- Redigest PRS416 and test plasmid with EcoRI and HindIII
Procedure:
- Run 7x8cm 50mL 1% agarose with 2.5uL EB gel
- Lanes: PRS X Ladder, PAR...6wells
Results:
- Gel Pics:


- Did it work?
- 4th colony section the best