 "
"
Team:Cornell/Description
From 2011.igem.org
(→DNA Assembly Methods) |
(→Light-Induced Lysis) |
||
| Line 510: | Line 510: | ||
=Light-Induced Lysis= | =Light-Induced Lysis= | ||
[[File:Cornell11_LightlysisDNA.png]] | [[File:Cornell11_LightlysisDNA.png]] | ||
| + | [[file:PZEBB1.png|320px|right]] | ||
<br> | <br> | ||
We would like to develop a genetic switch that is sensitive to specific wavelengths of visible light and use this gene expression system to lyse bacterial cells solely with light. The genetic light sensor is based on Chris Voigt’s "Multichromatic Control of Gene Expression in Escherichia coli," <sup>4</sup> which uses visible green light at 532 nm to induce specific gene expression. The system is composed of a light-activated surface protein, which autophosphorylates an intermediate chromophore and a reporter protein that binds to a specific promoter and is activated by the chromophore. | We would like to develop a genetic switch that is sensitive to specific wavelengths of visible light and use this gene expression system to lyse bacterial cells solely with light. The genetic light sensor is based on Chris Voigt’s "Multichromatic Control of Gene Expression in Escherichia coli," <sup>4</sup> which uses visible green light at 532 nm to induce specific gene expression. The system is composed of a light-activated surface protein, which autophosphorylates an intermediate chromophore and a reporter protein that binds to a specific promoter and is activated by the chromophore. | ||
Revision as of 03:46, 29 September 2011





Project Description |
Future Directions |
Business Development |
Outreach/HP |
Safety

Contents |
The BioFactory
Cornell’s BioFactory aims to develop a simple and efficient method for the construction of enzyme-immobilized surfaces capable of multi step chemical reactions.
As more chemical production techniques begin to utilize enzymatic reactions, genetic engineers must consider ways to resolve competing side reactions and the toxic accumulation of intermediates, reduce purification costs, and correctly express non-native enzymes or proteins in bacteria. In some cases, such challenges could be more easily resolved by simply extracting the molecular metabolic mechanisms and produce the target compound in a cell-free environment. We believe a cell-free system for biosynthesis can resolve these issues and still use the power of bacteria to help build devices capable of producing complex organic compounds.
Over the summer, Cornell's iGEM team engineered strains of E. Coli to produce modified enzymes from the biosynthetic pathway of Violacein which were immobilized on the surface of microfluidic devices and capable of converting an initial feed of substrate into prodeoxyvioalcein, a direct intermediate of the violacein pathway. The microfluidic chips were designed, built and tested in the lab using Cornell’s modern cleanroom and nanofabrication facilities. We additionally designed and began construction of a light-induced apoptosis system capable of lysing bacteria cultures, producing the necessary enzymes without the use of expensive reagents or extensive protocols.



Microfluidic Device
Overview:
In order to develop a scalable synthetic pathway the team required a means to apply enzymes
to proteins extracted from lysis.
This means would have to:
- Provide large enzyme exposure area within a small volume.
- Allow for scalability for industrial application.
- Be able to accommodate a system of separate enzymes as components of a chosen biosynthetic pathway.
- Be relatively simple and inexpensive
After a series of brainstorming and development sessions during the spring of 2011, the team
decided to move forward with the creation of a microfluidic device.
Concept Visualization (4/24/2011 - 5/1/2011):
Summary:
Initial concept developed. Visualized using Solidworks and Photoview 360.
Basis of Design:
Team Leader sketch. Conceptual model. Very basic knowledge of microfluidic systems.
Features:
a. Entire pathway present on single chip
b. Side channels used for dividing chip during coat process. Separate enzymes coated on separated sections.
Image:

|
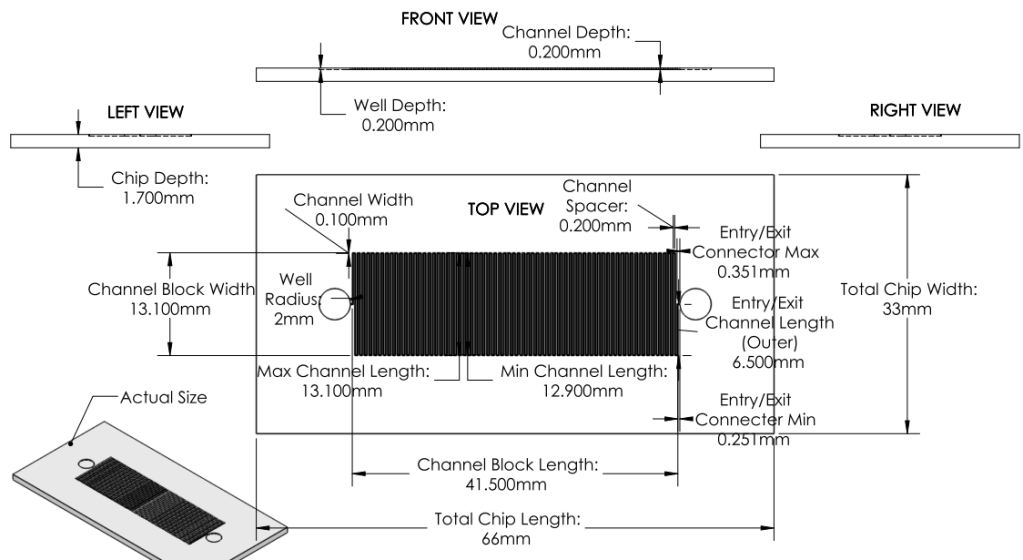
Revision 1: 1st Prototype Design (6/21/2011 - 6/27/2011):
Summary:
Formal microfluidics meeting. Developed effective modular system for biosynthetic pathway.
Design parameters based off of existing chip designs sold by MicroLiquid. (http://www.microliquid.com/microfluidic-products/chips/single-microfluidic-channel )
Using dimensions from MicroLiquid, optimized surface area using reasonable dimensions for channels.
Basis for Revision:
- Need for a concrete prototype design for critique and analysis.
- Greater knowledge of microfludics.
- Need for a prototype design applicable to the developed system.
Features:
System:
a. Entire pathway divided into several chips
- Each chip is coated with a single enzyme.
- Chips are linked together - substrate is flowed through linked chips to complete synthesis pathway.
Chip
- With sample specifications, maximize surface area to increase chances of successful synthesis process.
Image:
|

|
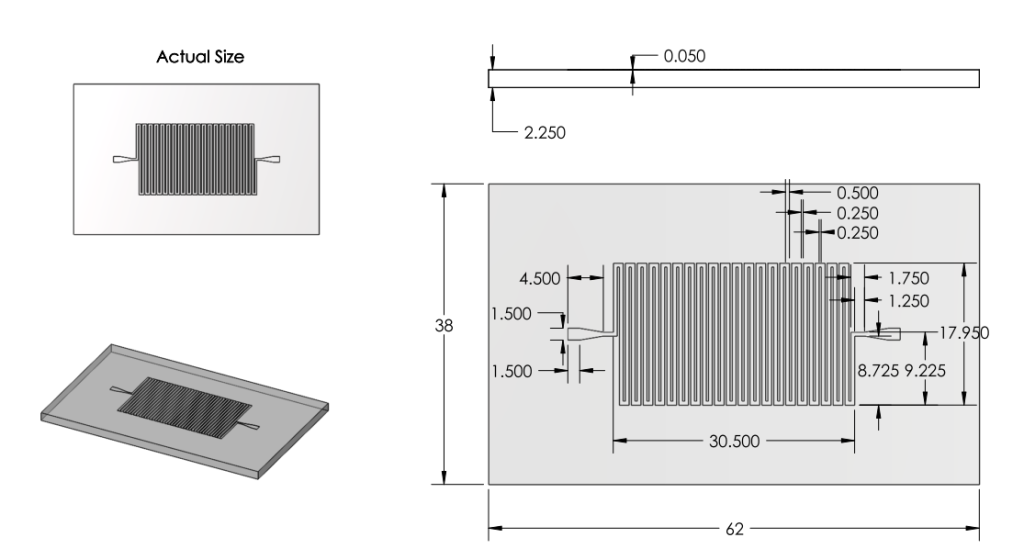
Revision 2: Modifications to Channels and Inlet/Outlets (6/27/2011 - 7/3/2011)
Summary:
Examined a variety of existing chips. Learned about more restrictions of mask printing.
Modified inlet and outlet shapes.
Also reduced number of channels, increased channel width and spacing in order to minimize impact of mask errors. New channel dimensions were based off of those from existing chips used for analyzing DNA.
Basis for Revision:
- Inlet and outlet round shape was unpractical given printing methods.
- Small channels with small spacing were prone to errors.
Features:
Inlet/Outlet
- Changed inlet/outlet shape to straight edge shapes.
Channel
Number
- Ran back of the envelope calculations for surface area. 22 turns of channel deemed satisfactory.
Dimensions
- Increased Channel width from 100microns to 500 microns.
- Decreased Channel Depth from 100 microns to 50 microns.
Overall Dimensions
- Reduced chip dimensions to reduce wasted space, keeping old margins.
|

|
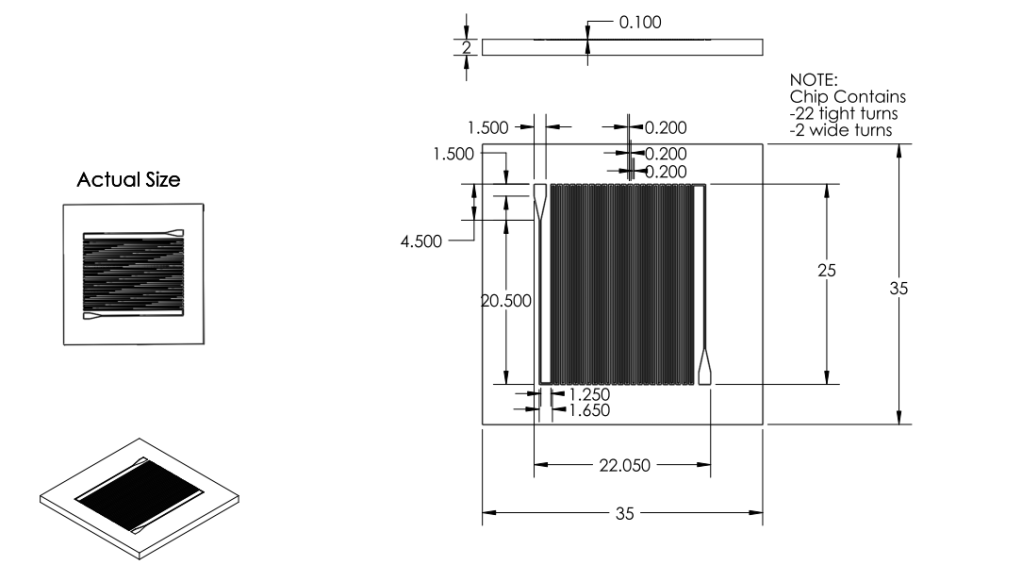
Revision 3: Final Chip Design (7/3/2011 - 7/6/2011)
Summary:
Optimized chip size to fit 4 chips on a 4'' diameter wafer.
After discussion, changed channel dimensions back to narrow safe limits of printing machine: 200 microns wide, 200 micron spacing.
Channel margins changed to limit of safe margin for wafer size.(1cm from edge)
Inlet/outlet positioning was changed to prevent channel damage when punching inlet/outlet holes.
Final Design was approved for printing.
Solidworks Sketch converted to DXF in Solidworks. Edited in Modo. Converted to GDS using LinkCAD and printed to mask.
Basis for Revision:
- Channels dimensions needed to balance space-savings and safe-print.
- Team wanted to print as many chips in as little space as possible.
- Team aimed to minimize damage when punching inlet/outlet holes.
Features:
Material:
We chose to create our microfluidic chip out of Polydimethylsiloxane
(PDMS) because of its extensive use in biologically related
microfluidic research. PDMS is an optically clear, nontoxic,
nonflammable and inert material that is ideal for working with
biological material. It is also a cost efficient material that can
easily be fabricated into a microfluidic device.
Inlet/Outlet
- Changed inlet/outlet position to reduce wasted space and reduce loss of chips
Channel
Dimensions
- Channel width changed to 200 microns.
- Channel spacing changed to 200 microns.
- Channel depth changed to 100 microns.
Overall Dimensions
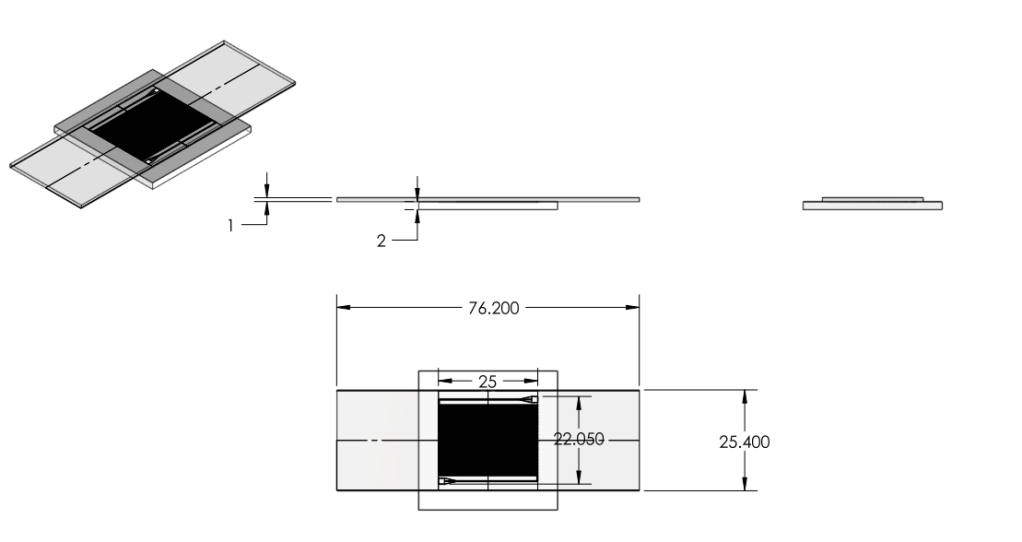
- Reduced chip print dimensions to 22.05mm by 25.00mm. Fits 4 safely on a 4'' diameter wafer. (101.6 mm diameter)
Mask
- Developed. Specifications drawing in Images below.
DXF to GDS File:

|
Images:
Chip:
|
|
Wafer:
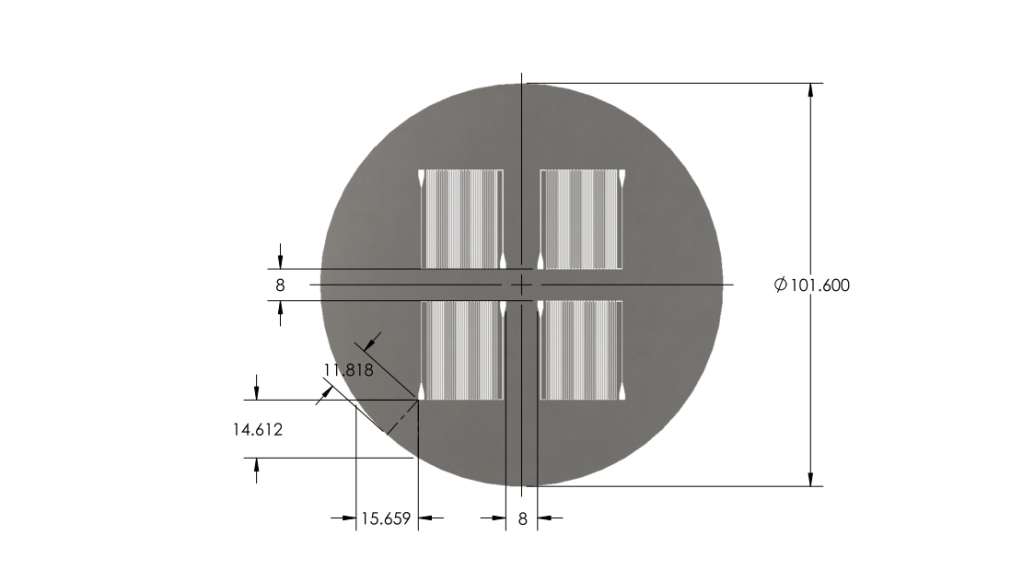
|

|

Process Summary:
We made use of the biotin-streptavidin binding interaction in fixing our biotinylated enzymes to the PDMS channel surfaces. E. coli has a natural mechanism encoded by the birA gene to produce biotin and biotinylate proteins. Thus, by avitagging enzymes of interest and adding biotin to the bacterial culture to enhance this natural mechanism, we functionalized our enzymes for biotin-streptavidin binding to the microfluidic device. We chose the biotin-avidin interaction over the competing nickel-histidine binding method commonly used for protein purification, as the avidin-biotin binding interaction lasts for longer.
Using standard chemical procedures from Gleghorn et al.1 provided by Dr. Kirby (Cornell University, Mechanical and Aerospace Engineering), NeutrAvidin was bonded to the surface of PDMS channels. NeutrAvidin is an avidin interchangeable with streptavidin, with a similar tetramer structure, which binds with biotin in four regions. NeutrAvidin can bind greater than 12 µg of biotin per mg protein and has a dissociation constant Kd = 10-15 M.2

Violacein Pathway

We chose to use the violacein pathway fully characterized by Balibar et. al.3; as our model enzyme-mediated-reaction. The violacein pathway involves five enzymes (VioA, VioB, VioC, VioD, and VioE) in the conversion of L-Tryptophan into violacein, a purple chromophore. This pathway was an especially attractive candidate for our project not only because it has been thoroughly characterized in E. coli, but also because relatively few enzymes are needed to convert a cheap, common substrate into a visualizable product. Furthermore, only three of the five enzymes (VioA, VioB, and VioE) are required to produce a colored product. Even with the exception of Vios C and D, L-Tryptophan is converted into prodeoxyviolacein, a green pigment (Figure 3A in Balibar et. al.3). Thus, we chose to biotinylate only VioA, VioB, and VioE to provide proof-of-concept that enzymes bound to our microfluidic devices may be used to facilitate enzyme-mediated reactions.
Violacein Pathway

Light-Induced Lysis


We would like to develop a genetic switch that is sensitive to specific wavelengths of visible light and use this gene expression system to lyse bacterial cells solely with light. The genetic light sensor is based on Chris Voigt’s "Multichromatic Control of Gene Expression in Escherichia coli," 4 which uses visible green light at 532 nm to induce specific gene expression. The system is composed of a light-activated surface protein, which autophosphorylates an intermediate chromophore and a reporter protein that binds to a specific promoter and is activated by the chromophore.
The genes to be expressed downstream of the promoter make up a lysis cassette derived from the lambda phage and developed by Prof. Young at Texas A&M University. This lysis system is very useful because the incubation period after gene expression is on the order of 50 minutes, and the actual lysis occurs within one minute thereafter. Our hope is to lyse bacterial cultures within a known timeframe and with specificity to green light.
DNA Assembly Methods
Site-Directed Mutagenesis
The Cornell iGEM team initially encountered problems expressing RFP and the VioA, B, and E enzymes. A possible reason for this issue may be excessive nucleotide spacing in between the ribosome binding site and the start codon in each of the protein-coding DNA sequences. More specifically, our constructs had thirty base pairs of separation, consisting of mainly the iGEM prefix and other restriction sites that we introduced for insertion into our pZE12 vector backbone. In contrast, greater gene expression may require shorter separation of eight to twelve base pairs.
QuikChange II site-directed mutagenesis is a method that can delete stretches of adjacent nucleotides. The team used this PCR-mediated technique to omit the iGEM prefix and the restriction sites necessary for insertion into vector solely to achieve protein expression and characterize the microfluidics chips.
In general, QuikChange II site-directed mutagenesis requires a vector of double-stranded DNA that contains an insert of interest. Two oligonucleotide primers are designed to have the desired mutation, which in this case is a deletion. The forward primer can be thought of as two internal parts. The first half is made of nucleotide sequence from the plasmid that lies to the left of the deletion site, while the second half reflects the vector sequence that is to the right of the deletion site. The reverse primer is simply the reverse complement of the forward primer. The purpose of the primers' divided mirroring of the vector nucleotide sequence is to make the deletion site loop out by means of matching base pairs flanking that site. Lastly, primer constructs should meet certain guidelines.
A high-fidelity DNA polymerase, such as PfuUltra, is necessary because it will be synthesizing the entire length of the vector backbone, while omitting the staggered nicks of the deletion site. Thus, the goal is to minimize the occurrences of a mutation during extension of the primers.
A complete PCR product of the vector, remade without the deletion site, does not contain methylated DNA. In contrast, parental forms of the vector and partially mutated backbones will exhibit the dam methylation that is common of DNA isolated from E. coli. Because DpnI recognizes the sequence (5' – GmATC – 3'), a digest with this restriction enzyme will select for only the deletion mutant vectors.
Annealed Primer Insertion
While there are multiple ways of preparing a gene for insertion into a vector, annealed oligonucleotides are especially convenient for introducing fragments such as promoters or polylinkers. Primer dimers bypass the need for digestion and PCR amplification of the insert. The specific construction of the primers makes for a simple insertion technique.
First, the two complementary oligos are staggered in design so that there are 5' and 3' overhangs representing the restriction site sequences of the digested vector backbone. Second, the primers must be phosphorylated using T4 polynucleotide kinase in order to be ligated into a CIAP-treated backbone.
The Cornell iGEM team used two sets of annealed primers to introduce the AviTag sequence, a stop codon, and the iGEM suffix into the pZE12 vector backbone. The first set of primers consists of a top single-stranded primer with the SphI restriction site as a 5' overhang and about half of the AviTag DNA sequence. The bottom half in this first set is the complementary strand. The second set of primers consists of a top single-stranded primer with the rest of the AviTag, a stop codon, the iGEM suffix, and the ClaI restriction site as a 3' overhang. For each primer set, the top and bottom primers are annealed to form the gene insert.
References
- 1. Gleghorn, J. P. et al. (2010). Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip, 10, 27-29. doi:10.1039/B917959C
- 2. Avidin and streptavidin conjugates. (n.d.). Retrieved from http://www.invitrogen.com/site/us/en/home/References/Molecular-Probes-The-Handbook/Antibodies-Avidins-Lectins-and-Related-Products/Avidin-Streptavidin-NeutrAvidin-and-CaptAvidin-Biotin-Binding-Proteins-and-Affinity-Matrices.html
- 3. Balibar, C. J. and Walsh, C. T. (2006). In Vitro Biosynthesis of Violacein from L-Tryptophan by the Enzymes VioA−E from Chromobacterium Violaceum. Biochemistry, 45(51), 15444-15457. doi:10.1021/bi061998z
- 4. Tabor, J. J., Levskaya, A., & Voight, C. A. (2010). Multichromatic Control of Gene Expression in Escherichia coli. Journal of Molecular Biology, 405(2),315-324. doi:10.1016/j.jmb.2010.10.038