 "
"
Team:DTU-Denmark/Notebook
From 2011.igem.org
(Difference between revisions)
Zzhaojingg (Talk | contribs) (→PChiP/PChiP(mutated)-lacZ fusion) |
|||
| (19 intermediate revisions not shown) | |||
| Line 1: | Line 1: | ||
{{:Team:DTU-Denmark/Templates/Standard_page_begin|Notebook}} | {{:Team:DTU-Denmark/Templates/Standard_page_begin|Notebook}} | ||
__TOC__ | __TOC__ | ||
| - | |||
== Constructed plasmids == | == Constructed plasmids == | ||
| - | |||
=== P<sub>ChiP</sub>/P<sub>ChiP(mutated)</sub>-lacZ fusion === | === P<sub>ChiP</sub>/P<sub>ChiP(mutated)</sub>-lacZ fusion === | ||
The plasmids containing this DNA fragments were obtained by the following procedure: | The plasmids containing this DNA fragments were obtained by the following procedure: | ||
| - | #'''PCR''' | + | #'''PCR''': |
| - | ##'''ChiP''' using E. coli w3110 genomic DNA as a template and TAQ enzyme. Using upstream part of ChiP and two different downstream primers (one wild type and one with mutation). we obtained two types of ChiP: original and mutated, which has altered binding site for ChiX. | + | ##'''ChiP:''' using E. coli w3110 genomic DNA as a template and TAQ enzyme. Using upstream part of ChiP and two different downstream primers (one wild type and one with mutation). we obtained two types of ChiP: '''ChiP original''' and '''ChiP mutated''', which has altered binding site for ChiX. |
| - | ##'''lacZ | + | ##'''lacZ:'''from BBa_I72005 biobrick using phusion enzyme. |
| - | #'''Restriction digestions''' | + | ##'''pSB1C3''' |
| + | #'''Restriction digestions''': | ||
##'''ChiP original''' and '''ChiP mutated''' with XbaI and NcoI, | ##'''ChiP original''' and '''ChiP mutated''' with XbaI and NcoI, | ||
| - | ##'''lacZ | + | ##'''lacZ''' with BspHI and PstI |
| - | + | ##'''pSB1C3''' with EcoRI and PstI [[File:DTU-chip-lacZ.png|300px|thumb|right| Fig.1. A linearized version of the P<sub>chiP</sub>-lacZ construct inside the pSB1C3 plasmid. Restriction sites used, as well as expected fragment sizes are indicated.]] | |
| - | ##'''pSB1C3''' with EcoRI and PstI | + | #'''Ligations''': |
| - | #''' | + | |
##'''ChiP normal''' with '''lacZ''' and '''pSB1C3''' | ##'''ChiP normal''' with '''lacZ''' and '''pSB1C3''' | ||
##'''ChiP mutated''' with '''lacZ''' and '''pSB1C3''' | ##'''ChiP mutated''' with '''lacZ''' and '''pSB1C3''' | ||
| - | #''' | + | #'''Transformations''' to ''E. coli'' NM522 cells. |
| - | #''' | + | #'''Plating the bacterial'''and grow over night in 37℃ incubator |
| - | #''' | + | #'''Start of a liquid culture''' of transformed strain. |
| - | #'''Mini prep the plasmid, do digestion analysis:'''with EcoRI, PstI, EcoRV. '''(fig.1)''' | + | #'''Mini prep the plasmid, do digestion analysis: '''with EcoRI, PstI, EcoRV. '''(fig.1)''' |
| - | + | #'''Transformation''' to NM522, IG9 and IG302 strain. | |
| - | #'''Transformation''' to NM522, IG9 | + | |
#'''Beta-Galactosidase Assay''' Pick 3 transformants of chip original and chip mutated, and grow. Make betagal assays +/- ara. | #'''Beta-Galactosidase Assay''' Pick 3 transformants of chip original and chip mutated, and grow. Make betagal assays +/- ara. | ||
=== P<sub>Tet</sub>-chiX fusion === | === P<sub>Tet</sub>-chiX fusion === | ||
The plasmids containing this DNA fragments were obtained by the following procedure: | The plasmids containing this DNA fragments were obtained by the following procedure: | ||
| - | #''' | + | #'''Start liquid culture of minigenes''': chiX, tetR |
#'''PCR''':pSB4k5 (or any other non cam non pSB1 pSB6 plasmid) | #'''PCR''':pSB4k5 (or any other non cam non pSB1 pSB6 plasmid) | ||
| - | #''' | + | #'''Purification of the PCR product''' |
| - | #''' | + | #'''Mini prep minigenes''' |
#'''Restriction digestions''': | #'''Restriction digestions''': | ||
##'''chiX''' with XbaI and PstI | ##'''chiX''' with XbaI and PstI | ||
| - | ##'''tetR''' with EcoRI and | + | ##'''tetR''' with EcoRI and SpeI |
##'''pSB1C3''' with EcoRI and PstI | ##'''pSB1C3''' with EcoRI and PstI | ||
##'''pSB4k5''' with EcoRI and PstI | ##'''pSB4k5''' with EcoRI and PstI | ||
| - | #''' | + | #'''Ligations''' of:'''tetR''' with '''chiX''' and '''pSB1C3''' |
| - | #''' | + | #'''Transformations''' to ''E. coli'' NM522 cells. |
| - | #''' | + | #'''Plating the bacterial'''and grow over night in 37℃ incubator |
| - | #''' | + | #'''Colony PCR'''with CS149/150 on transformants. '''gel analysis''' '''(fig.2)''' |
| - | #''' | + | #'''Start of a liquid culture''' of correct ones. |
#'''Mini prep the plasmid, do digestion analysis:'''with EcoRI, PstI [[File:DTU-ChiX.png|400px|thumb|right| Fig.2. Colony PCR of cells transformed with P<sub>tet</sub>-chiX inside pSB1C3, CS149 and CS150 primers were used. Expected band size is 1391 bp]] | #'''Mini prep the plasmid, do digestion analysis:'''with EcoRI, PstI [[File:DTU-ChiX.png|400px|thumb|right| Fig.2. Colony PCR of cells transformed with P<sub>tet</sub>-chiX inside pSB1C3, CS149 and CS150 primers were used. Expected band size is 1391 bp]] | ||
| - | #''' | + | #'''Subclone Ptet-chiX part into pSB4k5''' |
| - | #''' | + | #'''Transformation''' to ''E. coli'' NM522 cells. |
#'''Colony PCR''' with CS149/150 on transformants.Pick 4 positive colonies and grow overnight | #'''Colony PCR''' with CS149/150 on transformants.Pick 4 positive colonies and grow overnight | ||
| - | #'''Minipreps''' the corect ones, '''Transform''' into IG302/pSB1C3 | + | #'''Minipreps''' the corect ones, '''Transform''' into IG302/pSB1C3 PchiP-lacZ |
#'''Beta-Galactosidase Assay''' Pick 2 transformants and grow. Make Beta-Galactosidase Assay +/- atc. | #'''Beta-Galactosidase Assay''' Pick 2 transformants and grow. Make Beta-Galactosidase Assay +/- atc. | ||
=== P<sub>Bad</sub>-chiXR fusion === | === P<sub>Bad</sub>-chiXR fusion === | ||
The plasmids containing this DNA fragments were obtained by the following procedure: | The plasmids containing this DNA fragments were obtained by the following procedure: | ||
| - | #''' | + | #'''Start liquid culture of minigenes''': chiXR |
#'''PCR''': | #'''PCR''': | ||
##pSB3T5 (or any other non cam non pSB1 pSB6 plasmid) | ##pSB3T5 (or any other non cam non pSB1 pSB6 plasmid) | ||
##pBad | ##pBad | ||
##pSB1C3 | ##pSB1C3 | ||
| - | #''' | + | #'''Purification of the PCR product''' |
| - | #''' | + | #'''Mini prep minigenes''' |
#'''Restriction digestions''': | #'''Restriction digestions''': | ||
| - | ##'''pBad''' with | + | ##'''pBad''' with SpeI and EcoRI |
##'''chiXR''' with XbaI and PstI | ##'''chiXR''' with XbaI and PstI | ||
##'''pSB1C3''' with EcoRI and PstI | ##'''pSB1C3''' with EcoRI and PstI | ||
##'''pSB4k5''' with EcoRI and PstI | ##'''pSB4k5''' with EcoRI and PstI | ||
| - | #''' | + | #'''Ligations''' of:'''pBad''' with '''chiXR''' and '''pSB1C3''' |
| - | #''' | + | #'''Transformations''' to ''E. coli'' NM522 cells. |
| - | #''' | + | #'''Plating the bacterial''' and grow over night in 37℃ incubator |
| - | #''' | + | #'''Start liquid culture''' of transformed strain. |
#'''Mini prep the plasmid, do digestion analysis:'''with EcoRI, PstI '''(fig.3)''' [[File:DTU-ChiXR.png|370px|thumb|right| Fig.3. The gel picture of the P<sub>BAD</sub>-chiXR construct. The restriction enzymes used, and the sizes are indicated on the picture. EN = EcoRI + NcoI, EB = EcoRI + BamHI]] | #'''Mini prep the plasmid, do digestion analysis:'''with EcoRI, PstI '''(fig.3)''' [[File:DTU-ChiXR.png|370px|thumb|right| Fig.3. The gel picture of the P<sub>BAD</sub>-chiXR construct. The restriction enzymes used, and the sizes are indicated on the picture. EN = EcoRI + NcoI, EB = EcoRI + BamHI]] | ||
| - | #''' | + | #'''Subclone PBad-chiXR part into pSB3T5''' |
| - | #''' | + | #'''Transformation''' to ''E. coli'' NM522 cells. |
#'''Colony PCR''' with CS149/150 on transformants.Pick 4 positive colonies and grow overnight | #'''Colony PCR''' with CS149/150 on transformants.Pick 4 positive colonies and grow overnight | ||
#'''Minipreps''' the corect ones, '''Transform''' into NM522/pSB1C3PchiP-lacZ and IG09/pSB1C3PchiP-lacZ | #'''Minipreps''' the corect ones, '''Transform''' into NM522/pSB1C3PchiP-lacZ and IG09/pSB1C3PchiP-lacZ | ||
| Line 93: | Line 90: | ||
*CSlac downstream TCGTTGGGCTGATGATCATAA | *CSlac downstream TCGTTGGGCTGATGATCATAA | ||
| - | == P<sub>BAD</sub>/araC promoter | + | == Arabinose promoter improvement == |
| + | Biobrick BBa_I746908 containing P<sub>BAD</sub>/araC promoter, BBa_I0500, fused with GFP was the scaffold for improving and testing the promoter. | ||
| + | # First, set of primers was used to PCR the p<sub>BAD</sub> promoter. 5 primers annealing to the promoter, SRS56-1, -2, -3, -4, -SPL, introduced either designed changes in the -35 and -10 region (first 4 primers) or random point mutations in the spacer region (-SPL primter). The SRS57 primer was used in pair with each of the these primers. | ||
| + | # BBa_I746908 on the pSB1A2 plasmid was digested with BamHI and KpnI, while the PCR products were cut with BglII and KpnI. | ||
| + | # Plasmid digestion was SAP treated in order to lower the chance of self-religating of the plasmid during the ligation step. | ||
| + | # Now, the digested PCR products and the digested plasmid was ligated and transformed. | ||
| + | # Then, after purifying the plasmids, the constructs could be tested for the correct insert by digesting with KpnI and BamHI. If the correct constructs were created (PCR products inserted into the plasmid) then restriction with BamHI couldn't take place. This restriction site was lost when fused with fragment digested with BglII. As a result only one band could be observed on the gel picture. | ||
| + | # Once the constructs were confirmed we analysed the GFP expression in BioLektor with 0.2% arabinose as inducer. | ||
| + | Primers used: | ||
| + | *SRS56-1 tagcagatcttacctgacgctttttatcgcaactctctaTAAtttctccatacccgtttttttgggctagctactagag | ||
| + | *SRS56-2 tagcagatcttacTtgacActttttatcgcaactctctactgtttctccatacccgtttttttgggctagctactagag | ||
| + | *SRS56-3 tagcagatcttacctgacActttttatcgcaactctctaTAAtttctccatacccgtttttttgggctagctactagag | ||
| + | *SRS56-4 tagcagatcttacctgacgctttttatcgcaactctctacAgtttctccatacccgtttttttgggctagctactagag | ||
| + | *SRS56SPL tagcagatctNNNctgacgNNNNNNNNNNNNNNNNNNtactgtNNNtccatacccgtttttttgggctagctactagag | ||
| + | *SRS57 ccaacccgggtaacgagcaaagcactgaacacca | ||
{{:Team:DTU-Denmark/Templates/Standard_page_end}} | {{:Team:DTU-Denmark/Templates/Standard_page_end}} | ||
Latest revision as of 04:08, 22 September 2011
Notebook
Contents |
Constructed plasmids
PChiP/PChiP(mutated)-lacZ fusion
The plasmids containing this DNA fragments were obtained by the following procedure:
- PCR:
- ChiP: using E. coli w3110 genomic DNA as a template and TAQ enzyme. Using upstream part of ChiP and two different downstream primers (one wild type and one with mutation). we obtained two types of ChiP: ChiP original and ChiP mutated, which has altered binding site for ChiX.
- lacZ:from BBa_I72005 biobrick using phusion enzyme.
- pSB1C3
- Restriction digestions:
- ChiP original and ChiP mutated with XbaI and NcoI,
- lacZ with BspHI and PstI
- pSB1C3 with EcoRI and PstI
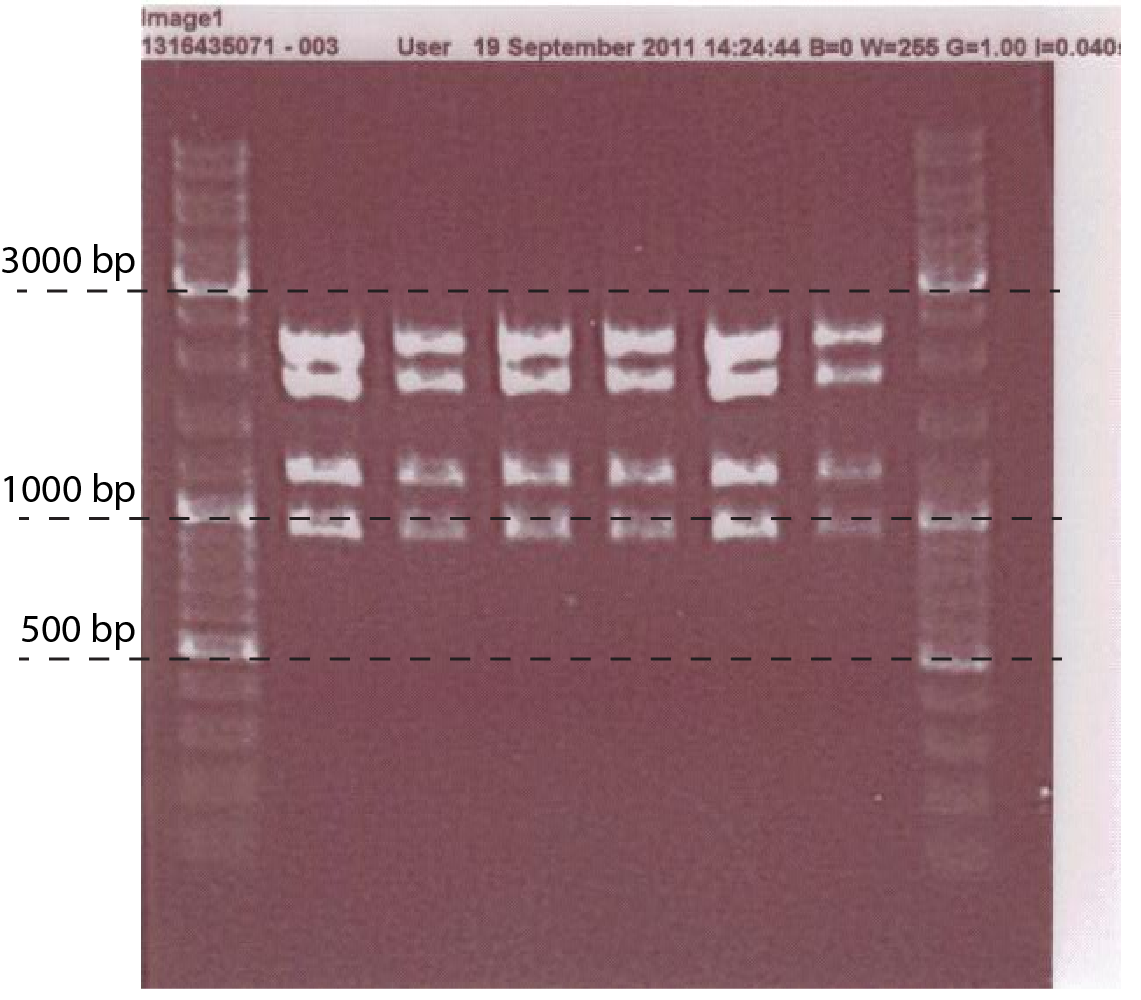 Fig.1. A linearized version of the PchiP-lacZ construct inside the pSB1C3 plasmid. Restriction sites used, as well as expected fragment sizes are indicated.
Fig.1. A linearized version of the PchiP-lacZ construct inside the pSB1C3 plasmid. Restriction sites used, as well as expected fragment sizes are indicated.
- Ligations:
- ChiP normal with lacZ and pSB1C3
- ChiP mutated with lacZ and pSB1C3
- Transformations to E. coli NM522 cells.
- Plating the bacterialand grow over night in 37℃ incubator
- Start of a liquid culture of transformed strain.
- Mini prep the plasmid, do digestion analysis: with EcoRI, PstI, EcoRV. (fig.1)
- Transformation to NM522, IG9 and IG302 strain.
- Beta-Galactosidase Assay Pick 3 transformants of chip original and chip mutated, and grow. Make betagal assays +/- ara.

PTet-chiX fusion
The plasmids containing this DNA fragments were obtained by the following procedure:
- Start liquid culture of minigenes: chiX, tetR
- PCR:pSB4k5 (or any other non cam non pSB1 pSB6 plasmid)
- Purification of the PCR product
- Mini prep minigenes
- Restriction digestions:
- chiX with XbaI and PstI
- tetR with EcoRI and SpeI
- pSB1C3 with EcoRI and PstI
- pSB4k5 with EcoRI and PstI
- Ligations of:tetR with chiX and pSB1C3
- Transformations to E. coli NM522 cells.
- Plating the bacterialand grow over night in 37℃ incubator
- Colony PCRwith CS149/150 on transformants. gel analysis (fig.2)
- Start of a liquid culture of correct ones.
- Mini prep the plasmid, do digestion analysis:with EcoRI, PstI
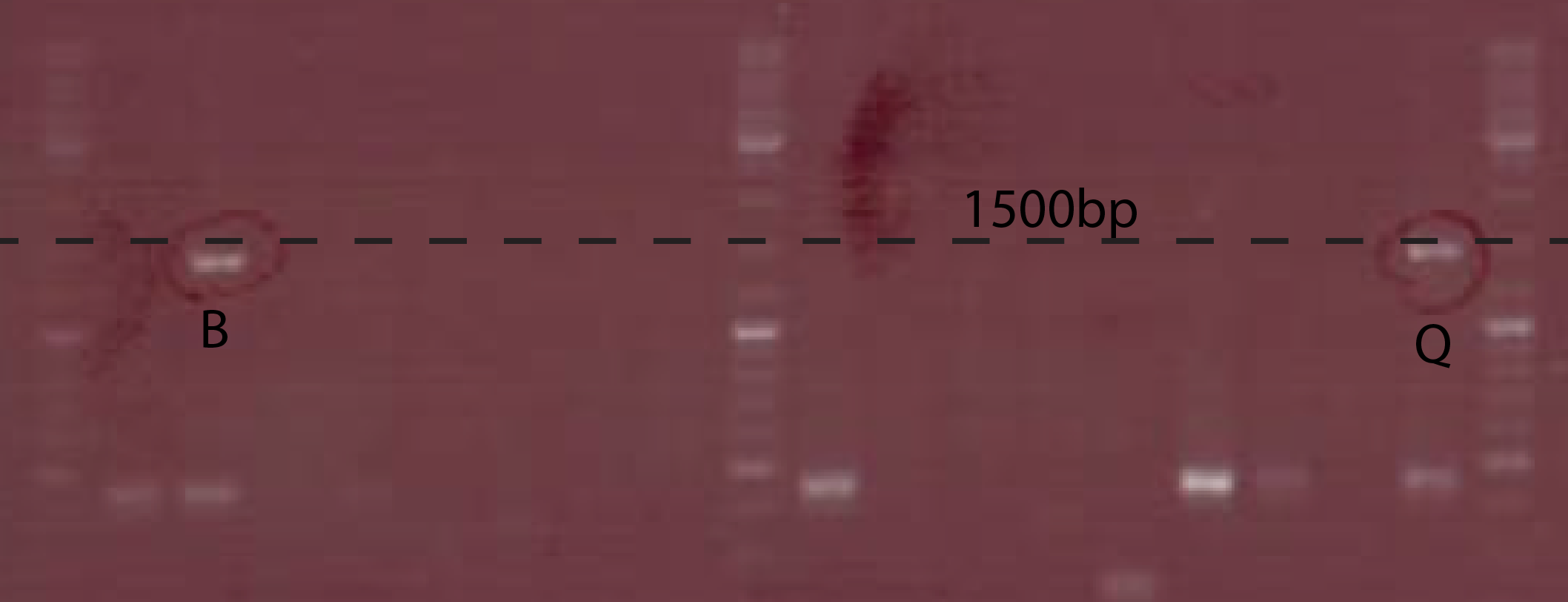 Fig.2. Colony PCR of cells transformed with Ptet-chiX inside pSB1C3, CS149 and CS150 primers were used. Expected band size is 1391 bp
Fig.2. Colony PCR of cells transformed with Ptet-chiX inside pSB1C3, CS149 and CS150 primers were used. Expected band size is 1391 bp - Subclone Ptet-chiX part into pSB4k5
- Transformation to E. coli NM522 cells.
- Colony PCR with CS149/150 on transformants.Pick 4 positive colonies and grow overnight
- Minipreps the corect ones, Transform into IG302/pSB1C3 PchiP-lacZ
- Beta-Galactosidase Assay Pick 2 transformants and grow. Make Beta-Galactosidase Assay +/- atc.

PBad-chiXR fusion
The plasmids containing this DNA fragments were obtained by the following procedure:
- Start liquid culture of minigenes: chiXR
- PCR:
- pSB3T5 (or any other non cam non pSB1 pSB6 plasmid)
- pBad
- pSB1C3
- Purification of the PCR product
- Mini prep minigenes
- Restriction digestions:
- pBad with SpeI and EcoRI
- chiXR with XbaI and PstI
- pSB1C3 with EcoRI and PstI
- pSB4k5 with EcoRI and PstI
- Ligations of:pBad with chiXR and pSB1C3
- Transformations to E. coli NM522 cells.
- Plating the bacterial and grow over night in 37℃ incubator
- Start liquid culture of transformed strain.
- Mini prep the plasmid, do digestion analysis:with EcoRI, PstI (fig.3)
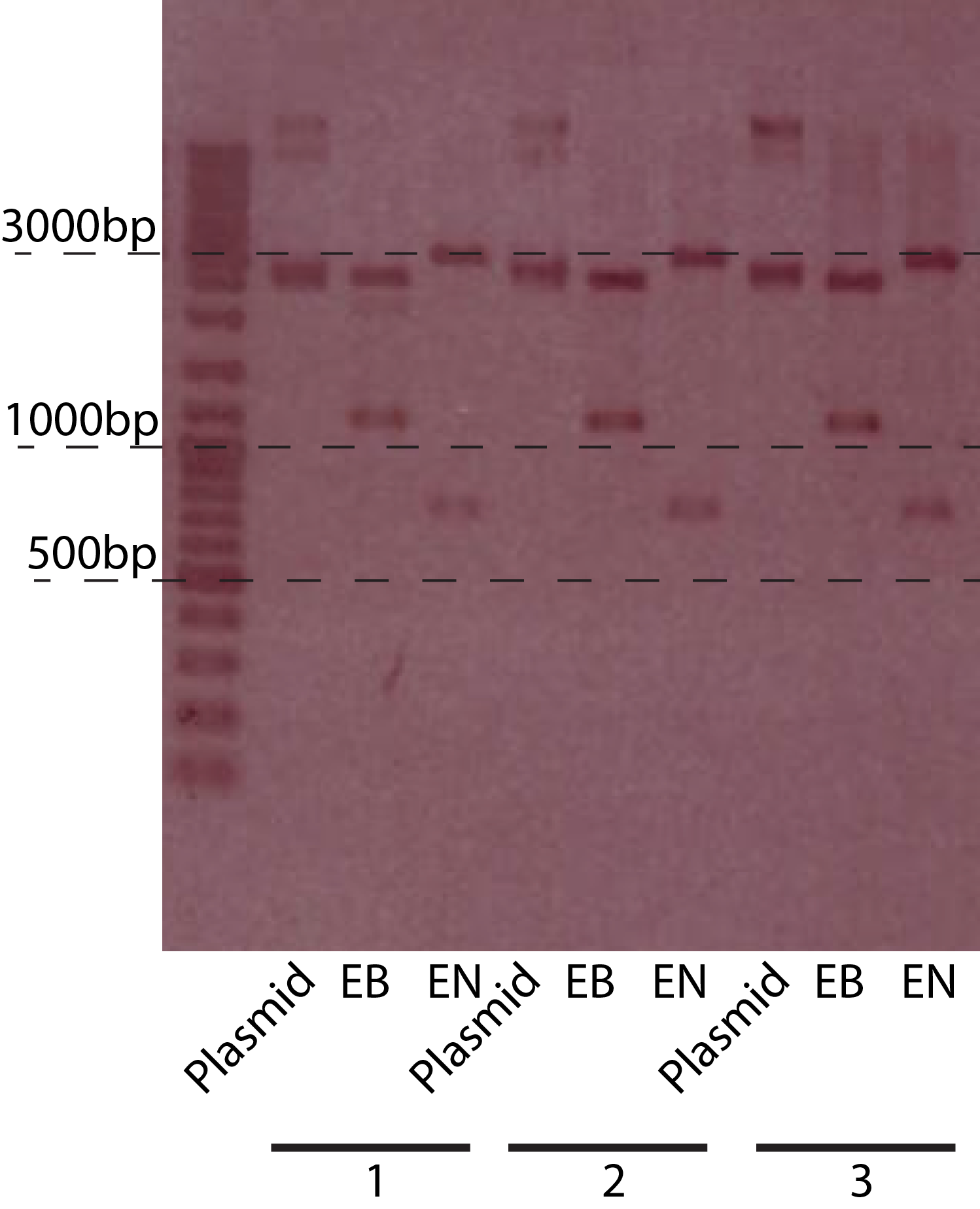 Fig.3. The gel picture of the PBAD-chiXR construct. The restriction enzymes used, and the sizes are indicated on the picture. EN = EcoRI + NcoI, EB = EcoRI + BamHI
Fig.3. The gel picture of the PBAD-chiXR construct. The restriction enzymes used, and the sizes are indicated on the picture. EN = EcoRI + NcoI, EB = EcoRI + BamHI - Subclone PBad-chiXR part into pSB3T5
- Transformation to E. coli NM522 cells.
- Colony PCR with CS149/150 on transformants.Pick 4 positive colonies and grow overnight
- Minipreps the corect ones, Transform into NM522/pSB1C3PchiP-lacZ and IG09/pSB1C3PchiP-lacZ
- Beta-Galactosidase Assay Pick 2 transformants and grow. Make betagal assays +/- ara.

Strain construction
The aim of this experimental section was to create strain with following gene deletetions:lacZYA and chbBCARG operon.
- PCR of recombineering linear pieces of DNA using pSLD31 plasmid and RS233/RS234 primers pair (lacZYA deletion) and chb upstream/downstream primers pair. pSLD31, was provided by our supervisor Sebastién Lemire, and it contains a kanamycin resistance gene flanked by lox66 and lox71 sites. The upstream/downstream primers pair anneal at the lox sites and they contain overhangs providing homology region to the sequences flanking "gene-to-be" deleted.
- Transformation of the E. coli W3110 strain containing pSLD18 plasmid (provides Red recombineering system) with pfhc3220 plasmid (containig Cre recombinase).
- Once the strain containing both plasmids was constructed we were ready to proceed with the first round of recombineering. We followed the protocol describing this procedure to achieve first gene deletion - $\Delta$chbBCARG. First we screened for the gain of kanamycin resistance and did the colony PCR to confirm correct insert size. Then the Cre recombinase was activated and facilitated pop-out event of the resistance marker. We screened for loss of resistance and, again, confirmed the correct sizes with colony PCR. Colony PCRs were done using CSchb upstream/downstream primers pair. Correct strain was saved as IG7 and stored under -80oC.
- As the next step, the IG7 strain was transformed with linear piece of DNA designed for lacZYA deletion. Gain of kanamycin resistance was screened for and the expected size of the insert was confirmed with colony PCR. After that the loss of resistance was conducted by growing the cells in minimum medium arabinose (0.15%). We screened for the pop-out event and colony PCRed the strain to confirm the correct recombineering. Primers used were called CSlac upstream/downstream. Finally, the strain was named IG9 and saved under -80oC.
Primers used:
- RS233 ACCAGTACGTTTTCCGCAGCTGTGGGTCAAAGAGGCATGACCTCCTTAGGAATTCTACCG
- RS234 GCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGACTAGCGGATCCTACCGTT
- chb upstream GGCCTGAGTTCTTAATTATCTTCGCGAATTATTTGCCCGATGTGTAGGCTGGAGCTGCTT
- chb downstream CCGGATGCAAGGTTCACGCCGCATCTGGCAAACATCCTCACATATGAATATCCTCCTTAG
- CSchb upstream TATCCGGCCTACAAAACTGTG
- CSchb downstream GCTGAATACGACACTGATCCAC
- CSlac upstream CGCTACAACGGGTAGCAA
- CSlac downstream TCGTTGGGCTGATGATCATAA
Arabinose promoter improvement
Biobrick BBa_I746908 containing PBAD/araC promoter, BBa_I0500, fused with GFP was the scaffold for improving and testing the promoter.
- First, set of primers was used to PCR the pBAD promoter. 5 primers annealing to the promoter, SRS56-1, -2, -3, -4, -SPL, introduced either designed changes in the -35 and -10 region (first 4 primers) or random point mutations in the spacer region (-SPL primter). The SRS57 primer was used in pair with each of the these primers.
- BBa_I746908 on the pSB1A2 plasmid was digested with BamHI and KpnI, while the PCR products were cut with BglII and KpnI.
- Plasmid digestion was SAP treated in order to lower the chance of self-religating of the plasmid during the ligation step.
- Now, the digested PCR products and the digested plasmid was ligated and transformed.
- Then, after purifying the plasmids, the constructs could be tested for the correct insert by digesting with KpnI and BamHI. If the correct constructs were created (PCR products inserted into the plasmid) then restriction with BamHI couldn't take place. This restriction site was lost when fused with fragment digested with BglII. As a result only one band could be observed on the gel picture.
- Once the constructs were confirmed we analysed the GFP expression in BioLektor with 0.2% arabinose as inducer.
Primers used:
- SRS56-1 tagcagatcttacctgacgctttttatcgcaactctctaTAAtttctccatacccgtttttttgggctagctactagag
- SRS56-2 tagcagatcttacTtgacActttttatcgcaactctctactgtttctccatacccgtttttttgggctagctactagag
- SRS56-3 tagcagatcttacctgacActttttatcgcaactctctaTAAtttctccatacccgtttttttgggctagctactagag
- SRS56-4 tagcagatcttacctgacgctttttatcgcaactctctacAgtttctccatacccgtttttttgggctagctactagag
- SRS56SPL tagcagatctNNNctgacgNNNNNNNNNNNNNNNNNNtactgtNNNtccatacccgtttttttgggctagctactagag
- SRS57 ccaacccgggtaacgagcaaagcactgaacacca
Sponsors
Thanks to:

