 "
"
Team:EPF-Lausanne/Notebook/July2011
From 2011.igem.org
(→J61002 plasmids) |
|||
| (100 intermediate revisions not shown) | |||
| Line 100: | Line 100: | ||
[[File:EPFL-08-07_igem_tetRvariants_1.jpg|thumb|right|Gel from the tetR mutants PCR. 1000kb ladder.]] | [[File:EPFL-08-07_igem_tetRvariants_1.jpg|thumb|right|Gel from the tetR mutants PCR. 1000kb ladder.]] | ||
| - | We ran the PCR on the tetR linear template, using Clara's primers for | + | We ran the PCR on the tetR linear template, using Clara's primers for PCR mutagenesis. |
Six PCRs were run. The first amplified the common sequence of the mutants: everything up to the mutated sites. The six other reactions amplified the second half of the gene, inducing specific mutations in tetR. | Six PCRs were run. The first amplified the common sequence of the mutants: everything up to the mutated sites. The six other reactions amplified the second half of the gene, inducing specific mutations in tetR. | ||
| Line 166: | Line 166: | ||
# YF42 | # YF42 | ||
# PQ39YM42 | # PQ39YM42 | ||
| - | # | + | # PQ39LV41YM42 |
The gel fluorescence traces show what appears to be PCR product in large amounts for the Common and EA37 reactions, and smaller amounts for the YF36 and YF42 reactions. The remaining three show almost no activity. | The gel fluorescence traces show what appears to be PCR product in large amounts for the Common and EA37 reactions, and smaller amounts for the YF36 and YF42 reactions. The remaining three show almost no activity. | ||
| Line 180: | Line 180: | ||
[[File:EPFL-2011-07-13_Stitch_PCR.jpg|thumb|First result from the fusion PCR: tetR mutant EA37 appears in "detectable" amounts]] | [[File:EPFL-2011-07-13_Stitch_PCR.jpg|thumb|First result from the fusion PCR: tetR mutant EA37 appears in "detectable" amounts]] | ||
| - | Douglas purified the gels containing yesterday's mutation-PCR products, following the | + | Douglas purified the gels containing yesterday's mutation-PCR products, following the [[Team:EPF-Lausanne/Protocols/Gel_purification| gel purification protocol]]. Isopropanol was added to the "Common PCR" product, as its expected length is inferior to 500bp. The final DNA concentrations, measured by photospectrometry, are listed in the following table. Even the very low concentration of YF36 should be sufficient for a PCR template, and the weird "260/280" ratio apparently results from agarose gel residues. |
{| | {| | ||
| Line 200: | Line 200: | ||
| - | These sequences were then used for a fusion PCR, to stitch mutants YF36, EA37, and YF42 to the common sequence, according to the protocol | + | These sequences were then used for a fusion PCR, to stitch mutants YF36, EA37, and YF42 to the common sequence, according to the [[Team:EPF-Lausanne/Protocols/TetR_Extension_PCR|overlap extension PCR protocol]]. Only the EA37 mutant, which was also the brightest on the PCR gels, yielded a significant amount of product. The next step is to check the product is the desired mutant, in order to confirm the success of this method. We also have to refine the PCR to get the other mutants to work; it seems the problem comes from the primers' melting temperature. |
---------------------------------------------------------------------------------- | ---------------------------------------------------------------------------------- | ||
| Line 222: | Line 222: | ||
=== pSB3C5 plasmid === | === pSB3C5 plasmid === | ||
| - | The PCR to add Pconst to TetR didn't work, but we already have it in the freezer. Nadine purified the pSB3C5 backbone, but the bands on the gel were really weak and the concentration is only 6 ng/ul. We will anyway try Gibson tomorrow... | + | |
| + | [[File:EPFL_Igem_1407_pSB3C5.jpg|200 px|thumb|right|pSB3C5 backbone purification, both bands contain the same product.]] | ||
| + | |||
| + | The PCR to add Pconst to TetR didn't work, but we already have it in the freezer. Nadine purified the pSB3C5 backbone, but the bands on the gel were really weak even with all the PCR products loaded onto the gel and the concentration is only 6 ng/ul. We will anyway try Gibson tomorrow... | ||
| + | |||
| + | === tetR mutants === | ||
| + | |||
| + | Douglas designed primers for site-specific mutagenesis of tetR, following the [[Team:EPF-Lausanne/Protocols/Site-specific_mutagenesis|protocol]]. We are testing this as another approach to inducing mutations. The primers were designed for the same mutations as attempted by extension PCR, i.e.: | ||
| + | |||
| + | # V36F | ||
| + | # E37A | ||
| + | # P39K | ||
| + | # Y42F | ||
| + | # P39Q_Y42M | ||
| + | # P39Q_L41V ''([26 July 2011] The Y42M mutation was left out here)'' | ||
| + | |||
| + | <s>By laziness</s> For convenience, the primer codes were changed to match those used by Agilent's primer design software. For example, V36F indicates a substitution of amino acid F for V in position 36. The primers are listed in this Google spreadsheet: https://spreadsheets.google.com/spreadsheet/ccc?key=0AvMKvyibLKKxdENDWkNLRV9ZSHhvVU45RHQ0bnNCWFE&hl=en_US | ||
| + | |||
| + | == Friday, 15 July 2011 == | ||
| + | The IPTG test gave us bad results (as expected due to data came from digestion): no increase in RFP and no decrease for the T4 lysis cassette. | ||
| + | Alessandro made miniprep and the digestion for the only red colony that he inoculated the day before. | ||
| + | The digestion gave a band of the expected size of the Gibson assembly (4428 bp) but we were expecting to see 2 bands instead we can see just 1; maybe one restriction site has been removed by the PCR or by the assembly. | ||
| + | [[File:EPFL2011_15_07_digestion.png|x300px|right|thumb|Gibson assembly from the red colony doesn't show 2 bands but just one band between 4 and 5 kb. Maybe one restriction site had been lost during the assembly.]] | ||
| + | |||
| + | |||
| + | Nadine made Gibson reactions for pSB3C5-pConst-TetR and for J61002-Ptet-LacI-Plac-RFP/lysis: | ||
| + | * pSB3C5: | ||
| + | ** pSB3C5 backbone (purified from gel yesterday) | ||
| + | ** pConst-TetR (the one Vincent had made for J23019 plasmid) | ||
| + | * J61002 Ptet-LacI Plac-RFP | ||
| + | ** J61002-Ptet backbone (3 ul instead of 4 ul last time) | ||
| + | ** LacI ssrA rrnB T (2x more than last time) | ||
| + | ** Plac-RFP (2x more than last time) | ||
| + | * J61002 Ptet-LacI Plac-lysis | ||
| + | ** J61002-Ptet backbone (3 ul instead of 4 ul last time) | ||
| + | ** LacI ssrA rrnB T (2x more than last time) | ||
| + | ** Plac-T4 lysis cassette (2x more than last time) | ||
| + | |||
| + | We hope that with less backbone we will have a smaller proportion of plasmids with only the J61002 backbone. | ||
| + | |||
| + | |||
| + | Douglas and Nadine ran a PCR to prepare another Gibson assembly: adding pConst-TetR on J61002 Ptet-RFP plasmid. | ||
| + | * backbone: | ||
| + | ** primers: J61002-plasmid-f + J61002-Plasmid-r | ||
| + | ** template: J61002 Ptet-RFP (we took colony 1) | ||
| + | * TetR: | ||
| + | ** primers: J61002 TetR-f + J61002 TetR-r | ||
| + | ** template: repressilator plasmid | ||
| + | Unfotunately, none of these 2 reactions worked because there was no band to see in the UV light... | ||
| + | |||
| + | == Monday, 18 July 2011 == | ||
| + | |||
| + | Alessandro made IPTG stocks and inoculated cells of the red colony (from glycerol stock, plasmid J61002-LacI-RFP) in order to use them the next day to perform the IPTG test. | ||
| + | |||
| + | |||
| + | [[File:EPFL_Igem_1807_tetr+bb.jpg|200 px|thumb|right|Nothing to see on the gel]] | ||
| + | Nadine transformed cells with: | ||
| + | * pSB3C5-pConst-TetR | ||
| + | * J61002 ptet-LacI pLac-RFP | ||
| + | * J61002 ptet-LacI pLac-lysis | ||
| + | and plated them on ampicillin or chloraphenicol dishes. We need to wait until Wednesday to be able to test these plasmids. | ||
| + | |||
| + | |||
| + | To prepare next Gibson, Nadine aslo re-did the PCR on J61002-Ptet backbone and on TetR. Details are as follows: | ||
| + | * backbone: | ||
| + | ** primers: J61002-plasmid-f + J61002-Plasmid-r | ||
| + | ** template: J61002 Ptet-RFP (this time colony 4) | ||
| + | * TetR: | ||
| + | ** primers: J61002 TetR-f + J61002 TetR-r | ||
| + | ** template: repressilator plasmid | ||
| + | We took 62°C for annealing step and 1min for extension step. But it didn't work... | ||
| + | I checked the primers features and no hairpin or dimers formation seems to be problematic (delta G's were -14 kcal/mol for the worse cases). Based on Henrike's advice, I checked the melting temperatures for only the parts of the primers that actually bind to template DNA and they are significantly lower... So Nadine ran a PCR with 42°C annealing step instead of 62°C, let's see tomorrow if it's better. | ||
| + | |||
| + | == Tuesday, 19 July 2011== | ||
| + | |||
| + | Alessandro started the IPTG test and the results will be available the next day. | ||
| + | |||
| + | The PCR with 42°C for annealing step didn't work either... We'll put this assembly aside, hoping that we will be able to insert TetR in others plasmids. | ||
| + | |||
| + | The Gibson assemblies made last Friday and transformed yesterday show disappointing results: | ||
| + | * J61002 Ptet-LacI Plac-RFP: only two colonies and they both are white | ||
| + | * J61002 Ptet-LacI Plac-lysis: no colonies | ||
| + | * pSB3C5 Pconst-TetR: a lot of red colonies, although they should not have RFP | ||
| + | For the J61002 assemblies, we will do the PCRs again to have fresh parts, and then try again. For pSB3C5 the results are really strange, as we don't think RFP should be included in the original plasmid. Also, Vincent's results for the miniprep of the original are suspiciously high (60 ng/ul is more than expected)so perhaps it's not the plasmid we think it is. We will then try to use pSB3K1 plasmid. | ||
| + | |||
| + | Nadine and Alessandro designed the plasmid for the new Gibson assembly to put P-const-TetR on a new backbone (pSB3K1) because the pSB3C5 didn't give the expected results (high DNA concentration while expected low copy number). | ||
| + | |||
| + | == Wednesday, 20 July 2011 == | ||
| + | |||
| + | [[File:EPFL2011_IPTG_test_results_20072011.PNG|thumb|The first image is the plot of the OD whereas the second is the plot of fluorescence of the RFP]] | ||
| + | |||
| + | The results from the IPTG test (picture below) show a slight difference in RFP fluorescence between the sample (w/ IPTG) and the negative control (w/ water) but this difference isn't significant therefore we must consider this as a bad result: we know our plasmid is not [[media:EPFL Plasmids(RFP instead of Lysis).png|as expected (the plasmid on the right)]], but we don't know what we have. | ||
| + | |||
| + | Alessandro made the transformation of the Repressilator plasmid because we haven't a sufficient amount to start PCRs. | ||
| + | Once we have that plasmid we can re-do PCRs to get the part for the Gibson assembly (RFP/Lysis plasmids). | ||
| + | Alessandro also designed the primers to do the colony-PCR wich we'll help us in a faster fashion to screen colony with the correct plasmid from the gibson assembly (instead of making minipreps and the following DNA digestion). | ||
| + | |||
| + | Douglas prepared agar-ampicillin plates for the mutagenesis, and transformed a pF3A plasmid with GFP (and ampicillin resistance). It can be used for a trial run of mutagenesis if the gibson assembled tetR plasmid is not yet available. He also transformed some repressilator, so we should have plenty to keep us going for a while. Also, Nadine debriefed him on her work for the past few weeks, so he can take over while she is on holiday. | ||
| + | |||
| + | == Thursday, 21 July 2011 == | ||
| + | |||
| + | Douglas and Alessandro inoculated the colonies transformed with the repressilator plasmid to make minipreps the following day. Alessandro also made the LB broth (because one bottle got contaminated) and distilled water. | ||
| + | |||
| + | [[File:EPFL-2011-07-21_Gibson_detector_fragments_PCR.jpg|thumb|right|Gel from our first attempt to replicate Nadine's PCR, to create fragments for the J61002-based reporter plasmid. None of the expected products show up: they should all be between 700 b and 2.4 kb]] | ||
| + | |||
| + | Douglas and Alessandro repeated Nadine's unsuccessful PCR, to make the '''fragments for Gibson assembly of the 'reporter plasmid' ''' (pTet-lacI-pLac-RFP/Lysis). The PCR was just as '''unsuccessful''', showing no trace of the expected products, as shown on the figure to the right. The fragments being assembled are: | ||
| + | |||
| + | * A J61002 '''backbone''' amplified from the J61002-pTet-RFP plasmid | ||
| + | * '''LacI''' amplified from the Repressilator plasmid | ||
| + | * '''pLac-RFP''' from the T4 Lysis plasmid | ||
| + | * OR '''pLac-T4Lysis''' also from the J61002-pTet-RFP plasmid | ||
| + | |||
| + | We found two similarly labelled tubes containing J61002-pTet-RFP, so we used both for our PCRs. The 'bis' reactions used the plasmid miniprepped by Allesandro, labelled in green writing. The 'non-bis' reactions used the other plasmid, of mysterious origin (miniprepped by Vincent perhaps?). | ||
| + | |||
| + | New reagents were prepared to repeat the PCR, using this time the optimal concentrations recommended by Bio-Rad for the iProof polymerase, i.e. primer concentrations of 0.5 uM and DNA template quantities between 1 pg and 10 ng for 50 ul. Specifically, we prepared a set with 1:1000 diluted template (resulting in a mass between 25 and 100 pg) and a set with 1:100 diluted template (resulting in a mass between 0.25 and 1 ng). They have been frozen with no polymerase, to attempt a touchdown PCR tomorrow. | ||
| + | |||
| + | == Friday, 22 July 2011 == | ||
| + | |||
| + | Alessandro made minipreps for the repressilator plasmid. | ||
| + | |||
| + | [[File:EPFL-2011-07-22_Gibson_TD_PCR_reporter.jpg|thumb|right|*EPIC FAIL* - Touchdown PCR to amplify the fragments for Gibson assembly of the two reporter plasmids]] | ||
| + | |||
| + | '''Touchdown PCR''' was run on yesterday's samples, to amplify the '''fragments for Gibson assembly of the reporter plasmids''', using the Bio-Rad thermal cycler. The thermal cycles were designed according to the iProof datasheet, using touchdown PCR recommendations from Korbie & Mattick (2008) [1]. Since the primer melting temperatures range from 47° C to 60° C, the cycle step the annealing temperature down from 65° C to 45° C over 20 cycles in constant decrements. It then repeats another 20 cycles at the 45° C floor temperature. Elongation lasts 40 s, following Bio-Rad's recommendation of 15 s/kb, for a 2400 kb product. The other parameters are as usual for iProof polymerase. | ||
| + | |||
| + | The PCR yielded absolutely '''no products'''. To further investigate the cause of failure, the PCR has been '''repeated''' using the Roche '''High Fidelity PLUS''' PCR kit, adapting the thermal cycles to the new polymerase. In a desparate move, the old PCR products were re-used for an almost-identical run on the Eppendorf cycler, with slightly longer elongation and denaturation steps (50 s and 7 s, respectively). | ||
| + | |||
| + | 1. Korbie, D.J. & Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature protocols 3, 1452-6(2008). | ||
| + | |||
| + | == Monday, 25 July 2011 == | ||
| + | |||
| + | [[File:EPFL-2011-07-25_Gibson_TD_PCR_HifiPLUS.jpg|thumb|right|Gibson fragments for the reporter plasmid, PCR'd with HifiPLUS enzyme. Good yield, but some unexpected product sizes, especially for the backbone.]] | ||
| + | |||
| + | Alessandro ran the Gel on Friday's PCR - that made Gibson fragments for the reporter plasmid, using the HifiPLUS enzyme. There is definitely some product (whereas iProof yields nothing at all). LacI and RFP seem to have worked properly. The lysis fragment is possibly a little short, and the backbone band is way too small. Overall, except for the backbone, it worked well. | ||
| + | |||
| + | In conclusion: our '''iProof enzyme seems not to work ''', either because it degraded, or it is just not suitable for this type of experiment. We should now use another enzyme. We ordered some High Fidelity PLUS for the next PCRs. | ||
| + | |||
| + | To investigate the lack of backbone product, Douglas ran a gradient PCR for the backbone fragment and the T4Lysis fragment. Five annealing temperatures were tested for each. The row temperatures are listed in the table below. To straddle their primers' expected annealing temperature, the backbone was placed in rows D--H and the Lysis fragment in rows A--E. Please note this PCR was run with the '''standard High Fidelity (not PLUS)''' enzyme! | ||
| + | |||
| + | {| | ||
| + | |+ Temperature settings for the gradient PCR | ||
| + | ! Row || Temperature [°C] | ||
| + | |- | ||
| + | | A || 65.0 | ||
| + | |- | ||
| + | | B || 63.8 | ||
| + | |- | ||
| + | | C || 61.6 | ||
| + | |- | ||
| + | | D || 57.6 | ||
| + | |- | ||
| + | | E || 52.7 | ||
| + | |- | ||
| + | | F || 48.9 | ||
| + | |- | ||
| + | | G || 46.2 | ||
| + | |- | ||
| + | | H || 45.0 | ||
| + | |} | ||
| + | |||
| + | == Tuesday, 26 July 2011 == | ||
| + | |||
| + | Alessandro made PCR to amplify pSB3K1 backbone to use for a gibson assembly to make the Pconst-TetR plasmid. We can't do the assembly now since we are waiting for primers for TetR. | ||
| + | The PCR gave good result for one colony (on two) as we can see a big band of the expected size (3.1 kb). | ||
| + | This time the purification will be performed not from the gel but directly from PCR with a commercial kit. | ||
| + | [[File:EPFL2011_pSB3K1_PCR_260711.jpg|thumb|right|pSB3K1 successfully amplified by PCR]] | ||
| + | Alessandro send the Vincent's plasmid (J6001-Ptet-RFP) to be sequenced (as the primer arrived). | ||
| + | ---------- | ||
| + | |||
| + | All the mutagenesis primers have been delivered, so Douglas diluted them. They are now all at 1 ug/ul concentrations, and need a further 1:10 or sodilution before being used for mutagenesis. He then ran a first mutagenesis on Alina's pF3A-tetR-GFP plasmid, introducing the following five mutations, in addition to a control reaction: | ||
| + | |||
| + | # V36F | ||
| + | # E37A | ||
| + | # P39K | ||
| + | # P39Q_Y42M | ||
| + | # P39Q_L41V | ||
| + | |||
| + | The mutagenesis kit (remains from last year) was almost empty, so he made 10 ul instead of 50 ul reactions. There should be enough remaining reagents for one more batch, possibly with control, but hardly any more. | ||
| + | |||
| + | The mutated plasmids were frozen over night, in order to digest and transform the following day. | ||
| + | |||
| + | ------------ | ||
| + | |||
| + | A gel was run for the gradient PCR, that used HiFi enzyme. It gave no product! In conclusion, we shall wait for the High Fidelity PLUS Enzyme, which should get here tomorrow. | ||
| + | |||
| + | In the mean time, Doug ran an identical gradient PCR with Matt's HiFiPLUS enzyme, changing the buffer concentration to 10 ul per 50 ul and lengthening the extension step to 2'30", as recommended by the datasheet. | ||
| + | |||
| + | == Wednesday, 27 July 2011 == | ||
| + | |||
| + | Alessandro purified the pSB3K1 backbone (to use for Gibson assembly and make the Pconst-TetR plasmid) from the PCR of the day before getting 60.6 ng/uL. | ||
| + | The sequencing reaction failed and checking the conditions of primers and template with NanoDrop, it seems like reverse primer has no DNA (but this doesn't explain why the reaction failed also with the forward primer); there's also a weird thing to annotate: checking the template on NanoDrop we always have an error of calibration. | ||
| + | Alessandro made new sample to send again sequencing. | ||
| + | |||
| + | --------------- | ||
| + | |||
| + | We '''FINALLY HAVE A WORKING PCR!!!!1111one''''. | ||
| + | |||
| + | [[File:EPFL-2011-07-27_Gibson_gradient_PCR_hifiPLUS.jpg|thumb|right|Backbone and Lysis fragments for the reporter plasmids, PCR'd with HifiPLUS. Expected product lengths: 2.4 kb backbone, 1.9 kb Lysis cassette.]] | ||
| + | |||
| + | More seriously, a gel was run on yesterday's gradient PCR products --- the PCR was ran using the High Fidelity PLUS enzyme, on the J61002-pTet backbone and pLac-T4Lysis fragments, for the Gibson assembly of the reporter plasmids. For both fragments, high quantities of specific product were yielded, though the backbone also contains large amounts of non-specific product. | ||
| + | |||
| + | For the backbone, lower annealing temperatures yield more product, as expected. The non-specific product seems relatively unaffected by annealing temperature (or it may decrease slightly at lower temperatures). The unspecific product present in large amounts is of similar length to the RFP gene, which is also present on the backbone template --- but determining its origin is unnecessary: it doesn't contain an antibiotic resistance gene, so plasmids assembled from it would not allow culture growth in the amplification step. | ||
| + | |||
| + | The T4 Lysis cassette fragment is a little too small. It could be due to GelRed affecting migration speed (according to Alina, it happens with large amounts of DNA), or it could indeed be an undesired product of shorter length. Please note the product quantities are barely affected by temperature (to the extent we can eyeball from the gel), whereas the expected primer melting temperatures are between 52° and 57°C, so we do not expect any product with 65°C annealing steps. | ||
| + | |||
| + | The assembly step was started. Transformation can be started tomorrow morning. | ||
| + | |||
| + | '''For the following PCRs''': Use Roche High Fidelity PLUS enzyme, and adapt any protocols to the appropriate specifications by Roche. Also, use either bottled water or the newer jar with fresher water (the one with autoclave tape on the lid). | ||
| + | |||
| + | == Thursday, 28 July 2011 == | ||
| + | |||
| + | The sequencing reaction this time went good therefore the plasmid is definitely the one expected. | ||
| + | Alessandro transformed and plated the plasmid to perform the day after the colony PCR just to optimize the protocol (this will be our positive control) for this technique that will be used to screen the gibson assembly. | ||
| + | |||
| + | Since seems to be some debris in the SOC medium bottle, we also plated it (under the same conditions of the cells) to test if something grows. Alessandro also tested the OD of the medium after 1 hour of incubation and no significant difference was found. | ||
| + | |||
| + | ------ | ||
| + | |||
| + | Douglas transformed and plated the Gibson-assembled reporter plasmids at the same time as Alessandro's plasmids. He later transformed it in a different ''E. Coli'' colony that over-expressed LacI, and therefore has a reduced probability of expressing the holin (from the lysis cassette) before they synthesise enough LacI to repress it. | ||
| + | |||
| + | Douglas also repeated the '''mutation PCR''' using the new High Fidelity PLUS enzymes. In a bold and dareful move, he programmed the thermal cycler to carry out both a touchdown PCR AND progressively extended elongation steps (during the second set of cycles, those at the floor annealing temperature, the elongation step is extended of five seconds every cycle). | ||
| + | |||
| + | == Friday, 29 July 2011 == | ||
| + | |||
| + | Douglas and Alessandro's transformed cells didn't grow (maybe problems with competent cells?). | ||
| + | Alessandro transformed the cells again (with the J61002-Ptet-RFP Gibson plasmid) but this time he trasformed also the sample labeled Col1-RFP and Col4-RFP (supposing that these are the same as J61002-Ptet-RFP Gibson plasmid). | ||
| + | Alessandro transformed the Gibson assembly made by Douglas on Lilia's (or Alina's?) competent cells and also on Henrike's competent cells. Due to an error only the lysis cassette has been transformed. On Tuesday the Gibson assembly has to be repeated since we ran out of the previous one. | ||
| + | |||
| + | The Mutation PCR yielded no product, probably because the primers were too diluted. It has been repeated with a higher primer concentration. | ||
{{:Team:EPF-Lausanne/Templates/Footer}} | {{:Team:EPF-Lausanne/Templates/Footer}} | ||
Latest revision as of 16:18, 2 August 2011
Notebook: July 2011
Friday, 1st of July 2011
Alina and Lilia did MITOMI on His-tetR expressed from linear template, it was loaded on chip in ITT expression mix. DNA was spotted on June 29 in different concentrations for both: consensus (tetO1) sequence and a random sequence ((-)control).

Images: Green fluorescence comes from green-lysine and red fluorescence comes from DNA (cy-5 labeled).
Although we don't observe much protein bound to the anti His-tag antibody, we can still see that it does bound TetO1 sequence and did not bind any (-)control sequence. Some possible reasons for low protein fluorescence under the buttons: the expression yield from the linear template with T7 promoter might be too low, the His-tag to antibody binding is not strong enough or the concentration of antibody is too low.
Friday, 1st of July 2011
Alina and Lilia amplified the His-tetR linear template. After PCR sample was loaded on 1% agarose gel with Rad Safe, “1Kb Plus DNA Ladder” was used.

We also prepared about thirty LB-agar plates with ampicilin, they are in the BM fridge.
Colonies from Gibson
After an overnight incubation, we found four colonies on the Ampicillin plates that had cells containing the plasmids from the Gibson assembly. The low number of colonies is attributed to the low competence of the cells. Of the four colonies, two were pink and two were yellow.
Tween experiment
Clara and Henrike tested the right dose of Tween 20 that can be used for the "chemostat" experiment without affecting the cell growth. Tween is a detergent that would prevent the cells from sticking and thus clogging the microfluidic channels. We tested 12 different dilutions of Tween 12 both for GFP and RFP e-coli which can be potentially used for the "chemostat" experiment. We read the OD(absorption @600nm) and fluorescence every 10 minutes, for 18 hours at 37°C.
Growth curves for all the different concentrations vs time


The legend shows the percentage of tween 20 that was added to the medium.
- We can observe that the growth factor is smaller for Chloramphenicol resistant e-coli due to the fact that Chloramphenicol is stronger than Kanamycin.
Calculated growth factor for the previous curves(Lilia)

Growth factor

We can observe that tween did not have a major effect on the cells' growth.
I suggest we use 0.075% tween in media for the experiments
Monday, 4th of July 2011
Alina and Vincent made plates (large and small) with chloramphenicol (concentration of 34 mg/mL). We used .74 mL of chloramphenicol for a liter of LB agar and plated them directly under the flame.
To figure out what was going wrong with the P23019 plasmid, we used the Nanodrop to determine that the sample (Plate 4, well 14E) was at a concentration of 97.4 ng/microL. This result seemed to indicate that there was DNA in the sample of the plate (as opposed to just red dye without DNA).
Tuesday, 5th of July 2011
Since we were running out of the P23019 plasmid DNA to try new PCRs, we thought it would be a good idea to transform it. We used both Lilia's and Alina's cells, adding a negative control. The incubation for chloramphenicol required a two hour recovery period in the incubator, since the antibiotic is stronger. After Tuesday's meeting, we went ahead and plated the result and let the plates incubate overnight.
Meanwhile, moving on with the Gibson assembly of the J61002 plasmid, Vincent tried to obtain the final Gibson sequence. This sequence would be used to see if PstI and SpeI would cut differently on the assembled plasmid versus the normal J61002 plasmid: we hoped this would allow us to check that Gibson had worked successfully if we ran the ligated plasmids on a gel. Ideally, the J61002 original plasmid would cut at two locations while the Gibson plasmid would only cut in a single location (the biobrick scar would make it impossible to cut at the previous cut location). On a gel, we would see a single piece of about 3000 bp for the Gibson plasmid and two bands of approximately 2200 and 700 bp respectively for the normal plasmid.
Using the plates with the four Gibson colonies, Vincent made cultures which he put in the incubator overnight.
Wednesday, 6th of July 2011
With the Gibson cultures in hand, Vincent made glycerol stocks (500 microL glycerol, 500 microL cells) and then mini-prepped the four cultures using the standard protocol. The resulting DNA plasmids were ready for enzyme ligase.
Vincent also looked at the results of the transformation of the P23019 using two different sets of competent cells and negative controls. No colonies grew, leaving us quite perplexed as to what was in the original P23019 well. Determined to find a substitute for the P23019 plasmid that would serve as a vector for the TetR sequence, we searched through the database looking for a plasmid with resistance to something other than ampicillin as well as a P15a origin of replication. We found four distinct possibilities, only two of which were available in the plates from the standard registry: pSB3K1 which has kanamycin resistance, and pSB3C5 which has chloramphenicol resistance (very similar to p23019).
Thursday, 7th of July 2011

Vincent and Nadine made a digestion on the minipreps from the 4 colonies that were transformed with the Gibson-extended J61002 plasmid. If Gibson was successful, only Pst1 will cut and we should have one band of 3094 bp. If Gibson didn't work, then Pst1 and Spe1 will cut and we should have 2 bands of 890 and 2060 bp. The digestion was made according to last year's protocol, with 1.5 hour incubation. The ladder on the gel is 1kb.
Colonies 1 and 4 have the expected band over 3kb, which is coherent with the observation made that they did express RFP. But colonies 2 and 3 have only one band... It is likely that in these cells the plasmid recombined in a shorter way, without RFP but with the resistance; otherwise the plasmid sequence is not what we think. We'll check tomorrow on the J61002 plasmid if we do get 2 bands on the starting plasmid. Also, we have some light smaller bands in colonies 1 and 4, indicating that the miniprep is perhaps not so pure.
Vincent and Alessandro transformed the J61002 original plasmid, since we were running out of DNA with which to compare to the Gibson assembly. In addition, they transformed the results of the previous day's registry search for convenient substitute plasmids (for p23019) using Alina's cells using 50 ng/microL concentration (Chloramphenicol Chl-SB3C5 70.6 ng/microL, K-SB3K1 67.7 ng/microL). These were plated appropriately and put in the incubator overnight.
Lilia and Alessandro did a MITOMI experiment on deBrujin sequences. Non of the sequences showed up to be bound nicely. But the protein pull down was not so robust due to the surface chemistry problems... Experiment needs to be repeated.


Friday, 8th of July 2011
Thanks to Henrike's expert eyes, we were able to harvest many colonies from the chloramphenicol pSB3C5 plasmid as well as many for the J61002 plasmid and the kanamycin plasmid. Since the chloramphenicol plasmid seemed closest to the p23019 plasmid for which we already had primers, it made sense to pursue the Gibson strategy for that plasmid. Alessandro and Vincent made culture tubes and put those in the incubator overnight. In the meanwhile, primers were designed for the next phase of Gibson assembly (i.e. the new pSB3C5 plasmid with the old TetR piece).

We ran the PCR on the tetR linear template, using Clara's primers for PCR mutagenesis.
Six PCRs were run. The first amplified the common sequence of the mutants: everything up to the mutated sites. The six other reactions amplified the second half of the gene, inducing specific mutations in tetR.
The fluorescence traces are insufficiently clear for accurate conclusions, therefore the PCR and gel will be repeated with different concentrations on Monday.
Saturday, 9th of July 2011
Vincent came in to pick up the J61002, pSB3C5, and pSB3K1 cultures from the incubator. He found that all the J61002 cultures were pink (as expected since they have the RFP gene). Both kanamycin cultures were pink, and two of the three chloramphenicol cultures were pink (though not as strong a pink as that of the J61002 plasmid). He went ahead and mini-prepped all the cultures, using hot water (55 C) instead of warm TE buffer during the elution step. He then made glycerol stocks (50% cell, 50% glycerol) and placed them in the -80 freezer. Finally, he measured the concentration of the resulting DNA after the mini-prep:
pSB3C5, colony 1: 35.2 ng/microL (accidentally used 2 microL, vs. 1 microL so more diluted)
pSB3C5, colony 2: 78.4 ng/microL
pSB3C5, colony 3: 70.2 ng/microL
pSB3K1, colony 1: 15.5 ng/microL
pSB3K1, colony 2: 14.3 ng/microL
J61002, colony 1: 54.2 ng/microL
J61002, colony 2: 74.3 ng/microL
J61002, colony 3: 45.9 ng/microL
Monday, 11th of July 2011
Nadine made a PCR to prepare next Gibson assembly, ran it on a gel and recovered the according fragments from the gel. Details of the 4 PCRs:
- ColE1 backbone with Ptet (from newly assembled J61002 plasmid), final concentration: 15.1 ng/ul
- LacI ssrA rrnB T on repressilator plasmid, final concentration: 46.7 ng/ul
- T4 lysis cassette on registry plasmid, final concentration: 29.7 ng/ul
- RFP amplification with Plac from Gibson J61002 plasmid, final concentration: 61,2 ng/ul

Alessandro made again a digestion on the 4 colonies recovered from last Gibson assembly, with this time also the original J61002 plasmid. See Thursday 7th July for more details. This confirms that colony 3 has somehow reassembled without RFP because it is smaller than colonies 1 and 4. The bands are bigger after digestion because the plasmids have become linear and they run slower than the coiled plasmids.

Tuesday, 12 July 2011
Douglas obtained satisfactory amounts of product from the mutation PCR on the tetR linear template. The most successful one required 1 uM of primers, mixed to the 100x diluted template (0.327 ng/uL concentration).


Again, seven PCRs were run. The first copies the common sequence, up to the mutated bases in the tetR coding region, with some padding (illustrated above). The next six copy the remaining sequence, and induce these specific mutations:
- VF 36
- EA37
- PK39
- YF42
- PQ39YM42
- PQ39LV41YM42
The gel fluorescence traces show what appears to be PCR product in large amounts for the Common and EA37 reactions, and smaller amounts for the YF36 and YF42 reactions. The remaining three show almost no activity.
The PCR products were subsequently separated using gel electrophoresis then cut out. They will be purified tomorrow.
Nadine made 2 Gibson assemblies with the PCR products form yesterday, transformed cells and plated them on ampicillin dishes. The newly assembled vectors will have ampicillin resistance, LacI under Ptet plus either lysis under Plac or RFP under Plac.
Wednesday, 13 July 2011

Douglas purified the gels containing yesterday's mutation-PCR products, following the gel purification protocol. Isopropanol was added to the "Common PCR" product, as its expected length is inferior to 500bp. The final DNA concentrations, measured by photospectrometry, are listed in the following table. Even the very low concentration of YF36 should be sufficient for a PCR template, and the weird "260/280" ratio apparently results from agarose gel residues.
| PCR # | Ref. | Conc. [ng/ul] | "260/280" |
|---|---|---|---|
| 1 | Common | 17.8 | 4.82 |
| 2 | YF36 | 1.9 | -0.26 |
| 2 (repeat) | YF36 | -4.4 | 0.36 |
| 3 | EA37 | 26.3 | 96.23 |
| 5 | YF42 | 39.4 | -5.24 |
| 5 (repeat) | YF42 | 40.1 | -2.41 |
These sequences were then used for a fusion PCR, to stitch mutants YF36, EA37, and YF42 to the common sequence, according to the overlap extension PCR protocol. Only the EA37 mutant, which was also the brightest on the PCR gels, yielded a significant amount of product. The next step is to check the product is the desired mutant, in order to confirm the success of this method. We also have to refine the PCR to get the other mutants to work; it seems the problem comes from the primers' melting temperature.
Alessandro and Nadine autoclaved beads and prepared 4 bottles of SOC medium. We took the plated cells (J61002 Ptet-LacI Plac-lysis & Ptet-LacI Plac-RFP) from incubator and put them in suspension (LB medium + ampicillin) to grow overnight. We also plated the rest of these transformed cells.
We did a Klenow reaction on the 1off library to have dsDNA. The ordered plasmids arrived,so we made a PCR on pSB3C5 (to amplify the backbone) and on TetR (adding pConst) to prepare the parts needed for Gibson.
Thursday, 14 July 2011
J61002 plasmids
Alessandro made minipreps on cells grown overnight and made glycerol stocks of them (500 microL). Because the cells that we assume to have RFP (under the control of Plac) are not a little red, we also kept an aliquot of these cells to make a test with IPTG (this is a molecular mimic of allocaltose therefore it triggers the transcription on Plac): if these cells become red with IPTG we can be sure that RFP is controlled by Plac in our plasmid (from Gibson assembly).
Alessandro digested the plasmind (from miniprep) with PstI because Nadine found that if the Gibson went well, they should have 2 restricition sites for this enzyme - instead of 1 for J61002 Ptet-RFP plasmid. Unfortunately, the results show that GIbson didn't work... For every colony we have a band at 2.5 kb, which corresponds exactly to the size of the backbone.

The backbone must have closed without the inserts... We can redo Gibson with less backbone. On the plates we prepared yesterday one colony is red! We suspended it, we'll check tomorrow if this one has the correct plasmid.
pSB3C5 plasmid

The PCR to add Pconst to TetR didn't work, but we already have it in the freezer. Nadine purified the pSB3C5 backbone, but the bands on the gel were really weak even with all the PCR products loaded onto the gel and the concentration is only 6 ng/ul. We will anyway try Gibson tomorrow...
tetR mutants
Douglas designed primers for site-specific mutagenesis of tetR, following the protocol. We are testing this as another approach to inducing mutations. The primers were designed for the same mutations as attempted by extension PCR, i.e.:
- V36F
- E37A
- P39K
- Y42F
- P39Q_Y42M
- P39Q_L41V ([26 July 2011] The Y42M mutation was left out here)
By laziness For convenience, the primer codes were changed to match those used by Agilent's primer design software. For example, V36F indicates a substitution of amino acid F for V in position 36. The primers are listed in this Google spreadsheet: https://spreadsheets.google.com/spreadsheet/ccc?key=0AvMKvyibLKKxdENDWkNLRV9ZSHhvVU45RHQ0bnNCWFE&hl=en_US
Friday, 15 July 2011
The IPTG test gave us bad results (as expected due to data came from digestion): no increase in RFP and no decrease for the T4 lysis cassette. Alessandro made miniprep and the digestion for the only red colony that he inoculated the day before. The digestion gave a band of the expected size of the Gibson assembly (4428 bp) but we were expecting to see 2 bands instead we can see just 1; maybe one restriction site has been removed by the PCR or by the assembly.

Nadine made Gibson reactions for pSB3C5-pConst-TetR and for J61002-Ptet-LacI-Plac-RFP/lysis:
- pSB3C5:
- pSB3C5 backbone (purified from gel yesterday)
- pConst-TetR (the one Vincent had made for J23019 plasmid)
- J61002 Ptet-LacI Plac-RFP
- J61002-Ptet backbone (3 ul instead of 4 ul last time)
- LacI ssrA rrnB T (2x more than last time)
- Plac-RFP (2x more than last time)
- J61002 Ptet-LacI Plac-lysis
- J61002-Ptet backbone (3 ul instead of 4 ul last time)
- LacI ssrA rrnB T (2x more than last time)
- Plac-T4 lysis cassette (2x more than last time)
We hope that with less backbone we will have a smaller proportion of plasmids with only the J61002 backbone.
Douglas and Nadine ran a PCR to prepare another Gibson assembly: adding pConst-TetR on J61002 Ptet-RFP plasmid.
- backbone:
- primers: J61002-plasmid-f + J61002-Plasmid-r
- template: J61002 Ptet-RFP (we took colony 1)
- TetR:
- primers: J61002 TetR-f + J61002 TetR-r
- template: repressilator plasmid
Unfotunately, none of these 2 reactions worked because there was no band to see in the UV light...
Monday, 18 July 2011
Alessandro made IPTG stocks and inoculated cells of the red colony (from glycerol stock, plasmid J61002-LacI-RFP) in order to use them the next day to perform the IPTG test.

Nadine transformed cells with:
- pSB3C5-pConst-TetR
- J61002 ptet-LacI pLac-RFP
- J61002 ptet-LacI pLac-lysis
and plated them on ampicillin or chloraphenicol dishes. We need to wait until Wednesday to be able to test these plasmids.
To prepare next Gibson, Nadine aslo re-did the PCR on J61002-Ptet backbone and on TetR. Details are as follows:
- backbone:
- primers: J61002-plasmid-f + J61002-Plasmid-r
- template: J61002 Ptet-RFP (this time colony 4)
- TetR:
- primers: J61002 TetR-f + J61002 TetR-r
- template: repressilator plasmid
We took 62°C for annealing step and 1min for extension step. But it didn't work... I checked the primers features and no hairpin or dimers formation seems to be problematic (delta G's were -14 kcal/mol for the worse cases). Based on Henrike's advice, I checked the melting temperatures for only the parts of the primers that actually bind to template DNA and they are significantly lower... So Nadine ran a PCR with 42°C annealing step instead of 62°C, let's see tomorrow if it's better.
Tuesday, 19 July 2011
Alessandro started the IPTG test and the results will be available the next day.
The PCR with 42°C for annealing step didn't work either... We'll put this assembly aside, hoping that we will be able to insert TetR in others plasmids.
The Gibson assemblies made last Friday and transformed yesterday show disappointing results:
- J61002 Ptet-LacI Plac-RFP: only two colonies and they both are white
- J61002 Ptet-LacI Plac-lysis: no colonies
- pSB3C5 Pconst-TetR: a lot of red colonies, although they should not have RFP
For the J61002 assemblies, we will do the PCRs again to have fresh parts, and then try again. For pSB3C5 the results are really strange, as we don't think RFP should be included in the original plasmid. Also, Vincent's results for the miniprep of the original are suspiciously high (60 ng/ul is more than expected)so perhaps it's not the plasmid we think it is. We will then try to use pSB3K1 plasmid.
Nadine and Alessandro designed the plasmid for the new Gibson assembly to put P-const-TetR on a new backbone (pSB3K1) because the pSB3C5 didn't give the expected results (high DNA concentration while expected low copy number).
Wednesday, 20 July 2011

The results from the IPTG test (picture below) show a slight difference in RFP fluorescence between the sample (w/ IPTG) and the negative control (w/ water) but this difference isn't significant therefore we must consider this as a bad result: we know our plasmid is not as expected (the plasmid on the right), but we don't know what we have.
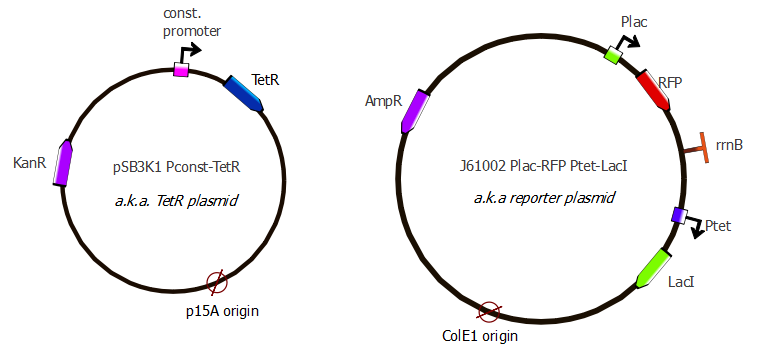{kind=link}
Alessandro made the transformation of the Repressilator plasmid because we haven't a sufficient amount to start PCRs. Once we have that plasmid we can re-do PCRs to get the part for the Gibson assembly (RFP/Lysis plasmids). Alessandro also designed the primers to do the colony-PCR wich we'll help us in a faster fashion to screen colony with the correct plasmid from the gibson assembly (instead of making minipreps and the following DNA digestion).
Douglas prepared agar-ampicillin plates for the mutagenesis, and transformed a pF3A plasmid with GFP (and ampicillin resistance). It can be used for a trial run of mutagenesis if the gibson assembled tetR plasmid is not yet available. He also transformed some repressilator, so we should have plenty to keep us going for a while. Also, Nadine debriefed him on her work for the past few weeks, so he can take over while she is on holiday.
Thursday, 21 July 2011
Douglas and Alessandro inoculated the colonies transformed with the repressilator plasmid to make minipreps the following day. Alessandro also made the LB broth (because one bottle got contaminated) and distilled water.

Douglas and Alessandro repeated Nadine's unsuccessful PCR, to make the fragments for Gibson assembly of the 'reporter plasmid' (pTet-lacI-pLac-RFP/Lysis). The PCR was just as unsuccessful, showing no trace of the expected products, as shown on the figure to the right. The fragments being assembled are:
- A J61002 backbone amplified from the J61002-pTet-RFP plasmid
- LacI amplified from the Repressilator plasmid
- pLac-RFP from the T4 Lysis plasmid
- OR pLac-T4Lysis also from the J61002-pTet-RFP plasmid
We found two similarly labelled tubes containing J61002-pTet-RFP, so we used both for our PCRs. The 'bis' reactions used the plasmid miniprepped by Allesandro, labelled in green writing. The 'non-bis' reactions used the other plasmid, of mysterious origin (miniprepped by Vincent perhaps?).
New reagents were prepared to repeat the PCR, using this time the optimal concentrations recommended by Bio-Rad for the iProof polymerase, i.e. primer concentrations of 0.5 uM and DNA template quantities between 1 pg and 10 ng for 50 ul. Specifically, we prepared a set with 1:1000 diluted template (resulting in a mass between 25 and 100 pg) and a set with 1:100 diluted template (resulting in a mass between 0.25 and 1 ng). They have been frozen with no polymerase, to attempt a touchdown PCR tomorrow.
Friday, 22 July 2011
Alessandro made minipreps for the repressilator plasmid.

Touchdown PCR was run on yesterday's samples, to amplify the fragments for Gibson assembly of the reporter plasmids, using the Bio-Rad thermal cycler. The thermal cycles were designed according to the iProof datasheet, using touchdown PCR recommendations from Korbie & Mattick (2008) [1]. Since the primer melting temperatures range from 47° C to 60° C, the cycle step the annealing temperature down from 65° C to 45° C over 20 cycles in constant decrements. It then repeats another 20 cycles at the 45° C floor temperature. Elongation lasts 40 s, following Bio-Rad's recommendation of 15 s/kb, for a 2400 kb product. The other parameters are as usual for iProof polymerase.
The PCR yielded absolutely no products. To further investigate the cause of failure, the PCR has been repeated using the Roche High Fidelity PLUS PCR kit, adapting the thermal cycles to the new polymerase. In a desparate move, the old PCR products were re-used for an almost-identical run on the Eppendorf cycler, with slightly longer elongation and denaturation steps (50 s and 7 s, respectively).
1. Korbie, D.J. & Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature protocols 3, 1452-6(2008).
Monday, 25 July 2011

Alessandro ran the Gel on Friday's PCR - that made Gibson fragments for the reporter plasmid, using the HifiPLUS enzyme. There is definitely some product (whereas iProof yields nothing at all). LacI and RFP seem to have worked properly. The lysis fragment is possibly a little short, and the backbone band is way too small. Overall, except for the backbone, it worked well.
In conclusion: our iProof enzyme seems not to work , either because it degraded, or it is just not suitable for this type of experiment. We should now use another enzyme. We ordered some High Fidelity PLUS for the next PCRs.
To investigate the lack of backbone product, Douglas ran a gradient PCR for the backbone fragment and the T4Lysis fragment. Five annealing temperatures were tested for each. The row temperatures are listed in the table below. To straddle their primers' expected annealing temperature, the backbone was placed in rows D--H and the Lysis fragment in rows A--E. Please note this PCR was run with the standard High Fidelity (not PLUS) enzyme!
| Row | Temperature [°C] |
|---|---|
| A | 65.0 |
| B | 63.8 |
| C | 61.6 |
| D | 57.6 |
| E | 52.7 |
| F | 48.9 |
| G | 46.2 |
| H | 45.0 |
Tuesday, 26 July 2011
Alessandro made PCR to amplify pSB3K1 backbone to use for a gibson assembly to make the Pconst-TetR plasmid. We can't do the assembly now since we are waiting for primers for TetR. The PCR gave good result for one colony (on two) as we can see a big band of the expected size (3.1 kb). This time the purification will be performed not from the gel but directly from PCR with a commercial kit.

Alessandro send the Vincent's plasmid (J6001-Ptet-RFP) to be sequenced (as the primer arrived).
All the mutagenesis primers have been delivered, so Douglas diluted them. They are now all at 1 ug/ul concentrations, and need a further 1:10 or sodilution before being used for mutagenesis. He then ran a first mutagenesis on Alina's pF3A-tetR-GFP plasmid, introducing the following five mutations, in addition to a control reaction:
- V36F
- E37A
- P39K
- P39Q_Y42M
- P39Q_L41V
The mutagenesis kit (remains from last year) was almost empty, so he made 10 ul instead of 50 ul reactions. There should be enough remaining reagents for one more batch, possibly with control, but hardly any more.
The mutated plasmids were frozen over night, in order to digest and transform the following day.
A gel was run for the gradient PCR, that used HiFi enzyme. It gave no product! In conclusion, we shall wait for the High Fidelity PLUS Enzyme, which should get here tomorrow.
In the mean time, Doug ran an identical gradient PCR with Matt's HiFiPLUS enzyme, changing the buffer concentration to 10 ul per 50 ul and lengthening the extension step to 2'30", as recommended by the datasheet.
Wednesday, 27 July 2011
Alessandro purified the pSB3K1 backbone (to use for Gibson assembly and make the Pconst-TetR plasmid) from the PCR of the day before getting 60.6 ng/uL. The sequencing reaction failed and checking the conditions of primers and template with NanoDrop, it seems like reverse primer has no DNA (but this doesn't explain why the reaction failed also with the forward primer); there's also a weird thing to annotate: checking the template on NanoDrop we always have an error of calibration. Alessandro made new sample to send again sequencing.
We FINALLY HAVE A WORKING PCR!!!!1111one'.

More seriously, a gel was run on yesterday's gradient PCR products --- the PCR was ran using the High Fidelity PLUS enzyme, on the J61002-pTet backbone and pLac-T4Lysis fragments, for the Gibson assembly of the reporter plasmids. For both fragments, high quantities of specific product were yielded, though the backbone also contains large amounts of non-specific product.
For the backbone, lower annealing temperatures yield more product, as expected. The non-specific product seems relatively unaffected by annealing temperature (or it may decrease slightly at lower temperatures). The unspecific product present in large amounts is of similar length to the RFP gene, which is also present on the backbone template --- but determining its origin is unnecessary: it doesn't contain an antibiotic resistance gene, so plasmids assembled from it would not allow culture growth in the amplification step.
The T4 Lysis cassette fragment is a little too small. It could be due to GelRed affecting migration speed (according to Alina, it happens with large amounts of DNA), or it could indeed be an undesired product of shorter length. Please note the product quantities are barely affected by temperature (to the extent we can eyeball from the gel), whereas the expected primer melting temperatures are between 52° and 57°C, so we do not expect any product with 65°C annealing steps.
The assembly step was started. Transformation can be started tomorrow morning.
For the following PCRs: Use Roche High Fidelity PLUS enzyme, and adapt any protocols to the appropriate specifications by Roche. Also, use either bottled water or the newer jar with fresher water (the one with autoclave tape on the lid).
Thursday, 28 July 2011
The sequencing reaction this time went good therefore the plasmid is definitely the one expected. Alessandro transformed and plated the plasmid to perform the day after the colony PCR just to optimize the protocol (this will be our positive control) for this technique that will be used to screen the gibson assembly.
Since seems to be some debris in the SOC medium bottle, we also plated it (under the same conditions of the cells) to test if something grows. Alessandro also tested the OD of the medium after 1 hour of incubation and no significant difference was found.
Douglas transformed and plated the Gibson-assembled reporter plasmids at the same time as Alessandro's plasmids. He later transformed it in a different E. Coli colony that over-expressed LacI, and therefore has a reduced probability of expressing the holin (from the lysis cassette) before they synthesise enough LacI to repress it.
Douglas also repeated the mutation PCR using the new High Fidelity PLUS enzymes. In a bold and dareful move, he programmed the thermal cycler to carry out both a touchdown PCR AND progressively extended elongation steps (during the second set of cycles, those at the floor annealing temperature, the elongation step is extended of five seconds every cycle).
Friday, 29 July 2011
Douglas and Alessandro's transformed cells didn't grow (maybe problems with competent cells?). Alessandro transformed the cells again (with the J61002-Ptet-RFP Gibson plasmid) but this time he trasformed also the sample labeled Col1-RFP and Col4-RFP (supposing that these are the same as J61002-Ptet-RFP Gibson plasmid). Alessandro transformed the Gibson assembly made by Douglas on Lilia's (or Alina's?) competent cells and also on Henrike's competent cells. Due to an error only the lysis cassette has been transformed. On Tuesday the Gibson assembly has to be repeated since we ran out of the previous one.
The Mutation PCR yielded no product, probably because the primers were too diluted. It has been repeated with a higher primer concentration.