 "
"
Team:BU Wellesley Software/Wet Lab
From 2011.igem.org

Wet Lab
Background
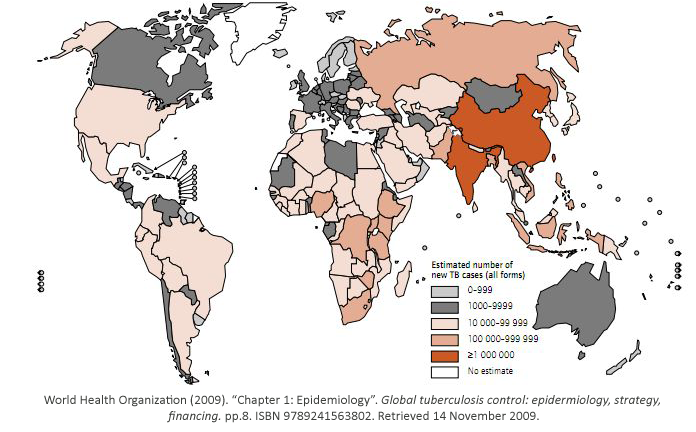
While some bacterial infections are seen as common place, others remain elusive to cure.
Tuberculosis has currently infected a third of the world’s population, and 1.7 million people
died from it in 2009. As shown, it is a worldwide problem that is prevalent in third world
countries.
Tuberculosis exists in two stages. In its latent stage, the immune system has it under
control and prevents the bacteria from reproducing. When it switches into its active stage as
a person's immune system becomes compromised, it begins to multiply and the person starts
showing symptoms of the disease.  The genetic mechanisms that trigger the change between the two
forms are not very well understood. The genetic network of tuberculosis is complicated and
studying the interactions between genes can be time consuming, requiring the construction of
many plasmids to study only one small part of the network. To look for new, faster ways of
studying this problem we turned to synthetic biology. Instead of creating many vectors
to study the interactions of genes, we aimed to create a plasmid with a designed circuit that
would control the transcription of several tuberculosis genes using invertases. We chose to
use non-pathogenic mycobacterium tuberculosis genes and transcription factors within non-
pathogenic E.Coli for safety reasons.
The genetic mechanisms that trigger the change between the two
forms are not very well understood. The genetic network of tuberculosis is complicated and
studying the interactions between genes can be time consuming, requiring the construction of
many plasmids to study only one small part of the network. To look for new, faster ways of
studying this problem we turned to synthetic biology. Instead of creating many vectors
to study the interactions of genes, we aimed to create a plasmid with a designed circuit that
would control the transcription of several tuberculosis genes using invertases. We chose to
use non-pathogenic mycobacterium tuberculosis genes and transcription factors within non-
pathogenic E.Coli for safety reasons.
Invertases are recombinases that will bind to recognition sites. Once bound, the DNA between two sites will invert and flip horizontally. This effectively acts as an on/off switch for transcription of that region of DNA as it is not available for DNA polymerase to bind to. In terms of circuit design, it will act as a not gate. Using them within a plasmid with several tuberculosis genes would allow us to check for regulatory measures such as negative feedback, etc.
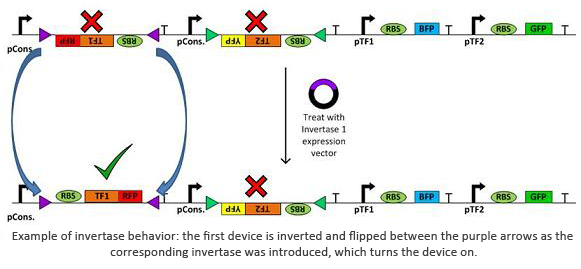
In addition to the novel cellular architecture, we also used a variety of software tools created by our computational teams (Boston University, Wellesley):
- G-nome Surfer Pro
G-nome Surfer Pro is an integrated environment that allows for the viewing of prokaryotic genomic data and literature associated with the genome. As it is built on a Microsoft Surface, it encourages collaboration. We used Optimus Primer on the G-nome Surfer to design the primers for the tuberculosis genes and transcription factors. - Trumpet
Trumpet consists of a library of genes and promoters, which can be configured into any desired permutation or combination by treating the DNA with recombinases, which allows us to rewire the genes and switches and study all their combinations. It was used by the wetlab to help us map out where the invertases should go in our plasmids in order to correctly turn on and off the segments of DNA we want to study. - PuppetShow
This suite includes a high level programming language for specifying biological protocols commonly used in the laboratory, which are then executed by a liquid-handling robot with minimal user intervention. We used this in conjunction with the robot (described below) to create samples that were simultaneously created manually to compare the results. - Clotho
Clotho is used to mange, create and store new biological building blocks in community based repositories. It includes a suite of tools that include PuppetShow and Trumpet, which were designed specifically for this project. Other tools that were used were:- Bull Trowel
- Spreadit Parts
- Spreadit Vectors
- Spreadit Features
- Optimus Primer-primer designer
- Feature Chomp-reads in APE and GENBANK files and takes the features found within the file to a feature database
- Batterboard-allowed us to electronically represent physical samples in the lab
Another way we looked into facilitating progress in studying genetic networks is through the use of automation:
- Robot The liquid handling robot was used to reproduce molecular biology protocols in a more efficient and accurate manner. The robot is controlled by Puppeteer which creates code necessary to choose the correct series of commands in EVOware,the robot's software.
- QIA Cube
Results
Being associated with a large computational team, the wetlab team was unique as our results not only consisted of creating novel forms of DNA, but evaluating the usefulness of the software tools our computational team produced. Our success was not only measured in the number of colonies we could get to grow, but also in how well we could incorporate these tools and give informative feedback.
The first vectors we were interested in creating were fluorescent protein reporters devices. Our goal was to eventually fuse genes with the reporter devices in order to establish that the genes were being turned on or off.It is important to understand the behavior of each promoter before using it in a complex plasmid architecture, because a baseline is needed to establish how it behaves independently of any additional transcription factors. In the wet lab, various devices were created with either constitutive or inducible promoters. The constitutive promoters like Bba_R2000, Bba_I14033, and Bba_R0040 cause the genetic devices to produce fluorescent protein at all times. But the inducible promoters like Bba_I13453 need a factor such as arabinose in order to promote the transcription of the fluorescent protein gene. Besides figuring out which promoters work best, we also wanted to create a small arsenal of different fluorescent proteins to use. Our devices include one of the following: red, yellow, green, and blue fluorescent proteins.
*TAILI CHART HERE PLEASE :)
The protocols we used to create these can be found '''here''' (link to list of protocols). They were used in a standard workflow shown below:
Shannon Diagram
We had two approaches in creating these devices: bottom up and top down. The bottom up approach consisted of manually combining each individual piece of the promoter+RBS+GENE+Terminator. Top down meant that we started with a composite part we found on the plates or isolated from another plasmid, usually consisting of a RBS+_FP+Terminator. Despite our plan to create a reconfigurable plasmid by the end of summer, we encountered some problems that prevented us from achieving that. In the first few days, we had an extremely low DNA quantification values for the plasmid. While the actual number of plasmid in the liquid culture may vary depending on whether the plasmid is a high or low copy, no DNA could be visualized in gel electrophoresis. In order to confirm if the correct bacteria grew in the liquid culture, a small sample was taken for gram staining and visualization under the microscope. It appeared that there were lots of other bacteria that got amplified, probably because the antibiotic selection did not work or our lab equipment got contaminated. So we autoclaved the pipet tips and prepared a new batch of antibiotic-equipped LB agar plates. Then, we had a problem with ligation where no colonies appeared on the transformed plates. We tried tinkering with the ligation protocols by using different insert to vector ratios, ethanol precipitations, and various ligation enzymes. We even tried treating the backbone with the CIP enzyme to prevent the backbone from ligating to itself. After several unsuccessful troubleshooting approaches, we tried pelleting the cell to increase the concentration of the comptent cells made and then attempted to use a commercial line of competent cell. Fortunately, the second method worked and we could build our genetic devices!
At first, we encountered a problem involving the failure to induce the pBad promoter. After adding various concentration of arabinose and using different protocols, it turned out that the commercial cell lines, which we used to solve the ligation problem, did not contain the AraC gene. Since the AraC gene has to exist and interact with arabinose to activate the pBad promoter, we then looked up the part that contains AraC gene and added it to the existing part. While no fluorescence was been observed under UV, the analysis of those samples with the FACS machine has produced an arabinose concentration versus fluorescence intensity data that showed significant increase in fluorescence intensity after the induction with arabinose.
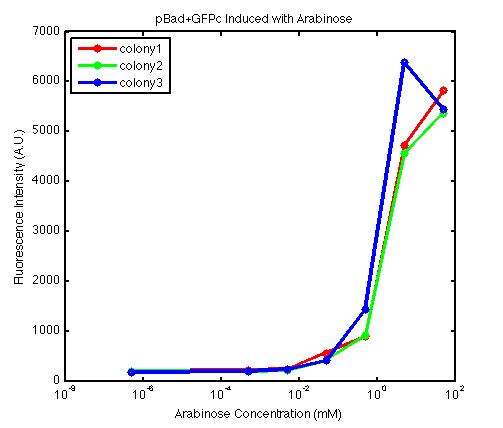
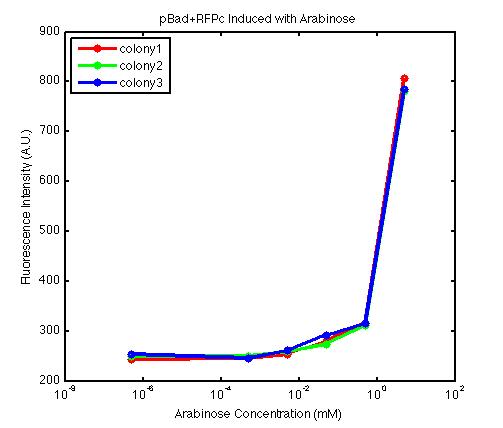
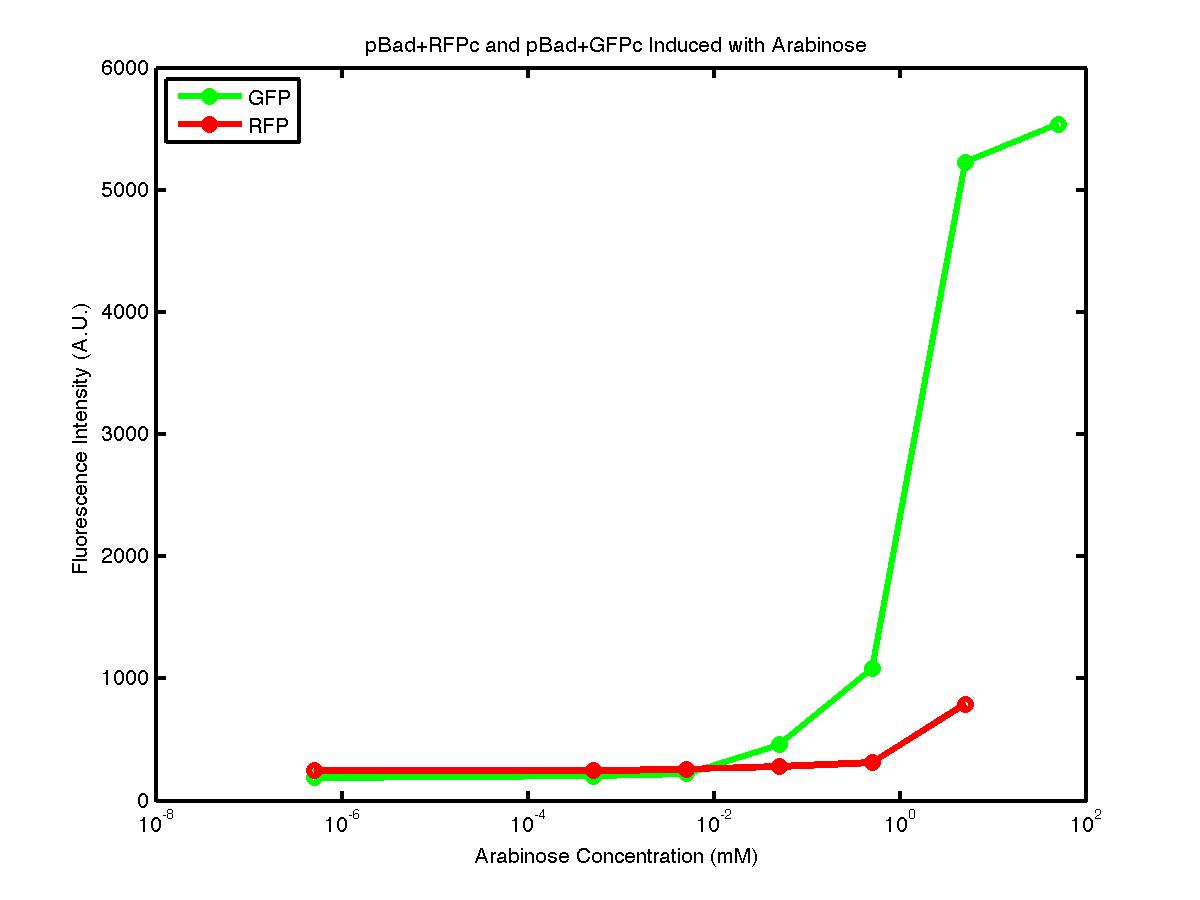
From the FACS data on arabinose induction and the resulting fluorescent protein gene expression, it appears that each genetic devices behave differently. While the tested device contain so much more components than just the reporter gene, but these constructs use a similar set of parts. Hence, the fluorescent intensity can be narrowed down enough to characterize the promoter and fluorescent genes. Before you continue reading, please note that fluorescent intensity uses arbitrary units so that such value is only meaningful when compared with other fluorescence signals.
The first graph shows the correlation between arabinose concentration and the fluorescence intensity expressed by the pBad+GFPc device. There were 3 colonies tested with the FACS machine and the plots of their expression levels show a relatively high degree of consistency. It appears that the effect of arabinose addition starts to manifest when the concentration of arabinose used is somewhat larger than 0.01 mM. If arabinose is added with a concentration that is one order of magnitude larger, the increase in fluorescence becomes drastically steeper up until the x value reaches ~5 mM. Then further addition will keep on increasing the fluorescence intensity, albeit with a smaller effect. The plot from the third colony is a bit unusual because it shows a more drastic increase, before falling down at the x value of ~5 mM. The huge gap between the last points from the first 3 points allow us to determine that ~5500 points difference in fluorescence intensity can be used to define an activated pBad+GFPc device when 50 mM arabinose is used.
The second graph shows the high correlation between arabinose concentration and the fluorescence intensity produced by the pBad+RFPc device. The 3 plot lines come from the 3 colonies tested with the FACS machine and the huge overlap between the lines implies a consistent pattern of gene expression. In this case, a visible gene expression occurs when the arabinose added has a concentration of 0.001 mM or higher. If arabinose is added with a concentration that is one order of magnitude larger, the increase in fluorescence becomes drastically steeper up until the x value reaches ~5 mM. Unlike the previous graph, the data does not tell us what will happen if arabinose with concentration higher than 5mM is used. The huge gap between the last points from the first 2 points allow us to determine that ~500 points difference in fluorescence intensity can be used to define an activated pBad+RFPc device when 5 mM arabinose is used.
By combining the running averages from the previous two graphs into the third graph, several conclusions can be reached on how the 2 devices compare. First, arabinose with concentration 0.001 mM or lower does not seem to induce any gene expression. Second, a similar pattern of induction can be observed in the individual behavior of pBAD+GFPc and/or pBad+RFPc. Nevertheless, direct comparison between them shows a great discrepancy between their intensity values. The expression level from the pBad+GFPc device reaches a peak value of ~5500, about one order of magnitude higher than the ~500 value from the pBad+RFPc device. In case both devices are incorporated into larger plasmid architecture, their relative behavior has to be noted as a baseline while analyzing the overall gene expression. Furthermore, a fluorescent intensity plot that appears flat when juxtaposed with another plot using the same axis does not prevent the possibility that they have the same characteristics that only differs in the relative values. In the end, it is important to remember that the highest expression with 50 mM arabinose is not even visible unless the FACS machine is used. Hence, an instrument with high sensitivity is needed to determine the activity of our genetic devices.
Approach
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Quisque ut tincidunt quam. Praesent in sodales mi. In hac habitasse platea dictumst. Nunc magna massa, gravida in euismod nec, rutrum sed dolor. Sed accumsan ullamcorper quam nec feugiat. Nam interdum volutpat nibh nec ultrices. Pellentesque habitant morbi tristique senectus et netus et malesuada fames ac turpis egestas. Donec eget nibh leo. Nullam euismod vulputate leo, eu egestas dolor volutpat at. Vivamus ac purus mi, vitae consequat enim.