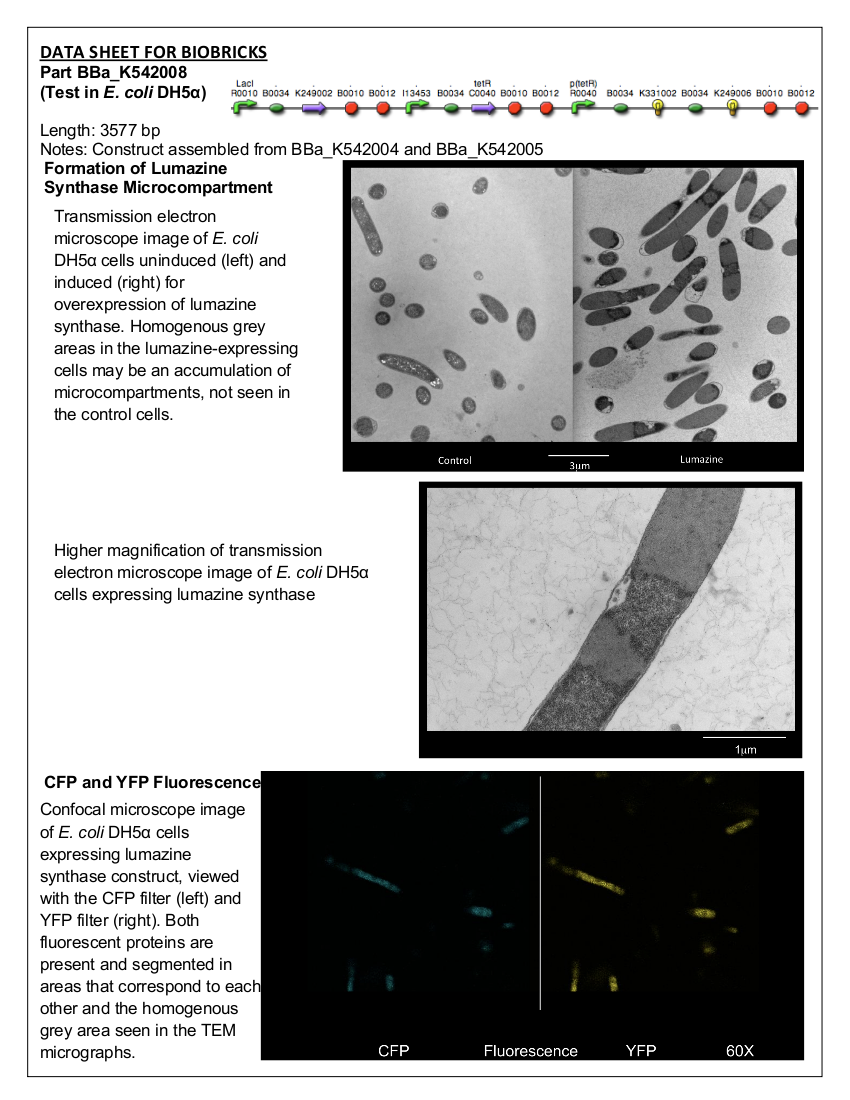
Part:BBa_K542008 – pLacI Regulated Lumazine Synthase and pBAD Inverse-Regulated Arg-tagged ECFP and EYFP – Test construct for a Lumazine Synthase microcompartment device
Datasheet for Part:BBa_K542008 in E. coli strain DH5alpha.
Introduction
The intended purpose of the Lumazine Synthase microcompartment device is to specifically target proteins that have been tagged with a positively charged oligopeptide into the cavity formed by the microcompartment. Furthermore, multiple unique proteins may be targeted into the cavity (1) for the purposes of increasing the efficiency of a metabolic pathway, to name just one example. This test construct is designed to demonstrate that proteins with positively charged tags can be localized into the cavity of the microcompartment formed by the oligomerization of Lumazine Synthase monomers.
The expression of Lumazine Synthase and the fluorescent proteins are independently regulated by two separate promoters. Lumazine Synthase is regulated by the pLacI promoter and the fluorescent proteins are regulated by the pBAD inverter. Since the two are independently controlled, Lumazine Synthase microcompartments may be formed in the presence or absence of the fluorescent proteins (ie. absence or presence of arabinose, respectively).
Because the fluorescent proteins are tagged with a positively-charged poly-arginine tag and the Lumazine Synthase is mutated with a negative interior (BBa_K249002 and Lethbridge 2009 Modeling), enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP) should be targeted into the microcompartments. ECFP and EYFP are a well known FRET pair.Construct
Part Number BBa_K542008
The construct was made by assembling BBa_K542004 with BBa_K542005.
Figure 1. BBa_K542008. This construct contains pLacI Regulated Lumazine Synthase and pBAD Inverse-Regulated Arg-tagged ECFP and EYFP.Characterization
Electron Microscopy
E. coli DH5α cells expressing BBa_K542008 and controls were viewed using Transmission Electron Microscopy (TEM).
Materials and Methods
E. coli DH5α cells containing BBa_K542008 as well as control cultures containing only plasmid pSB1C3 were grown overnight at 37°C. Approximately 400 mL of these cells were then resuspended in 5 mL SOC media, and induced with 5 mL or 10 mL of 1 M IPTG. Cells were again incubated at 37°C for approximately 4 hours. Cells were then spun down at 6000 x g for 2 min.
Cells were resuspended in 500 mL gluteraldehyde/formaldehyde fixative and incubated at room temperature for 2 hours. The solution was then spun at 6000 x g for 2 min.
The cell pellet was resuspended in 0.1 M sodium cacodylate buffer and incubated for 10 min. The solution was centrifuged for 2 min at 6000 x g. This step was repeated an additional 2 times. Cells were then resuspended in 1% osmium tetroxide fixative in 0.1 M CaC buffer and incubated at room temperature for 1 hour. The solution was centrifuged at 6000 x g for 2 min. Cells were then resuspended in MilliQ H2O.
The cells were spun down at 10000 x g for 5 min, and resuspended in 4% BIO-RAD standard low melting temperature agarose at 60°C. A drop of this cell-agarose mixture was then added to slides in order to harden, and incubated in MilliQ H2O overnight.
Agarose-cell mixtures were cut into cubes of approximately 1 mm x 1 mm x 1 mm. They were then incubated in increasing alcohol concentrations up to 100% anhydrous ethyl alcohol. The cubes were then incubated in decreasing concentrations of alcohol and increasing concentrations of resin, up to 100% resin. The cubes were left in 100% resin to harden overnight.
The resin-embedded cubes of cells were thin-sectioned and placed on a carbon grid. They were stained with uranyl acetate and then positively stained with lead citrate and viewed under TEM.Results
Figure 2. E. coli DH5α control cells (left) and cells expressing BBa_K542008 (right) as viewed under transmission electron microscope.
Figure 3. E. coli DH5α cells expressing BBa_K542008 as viewed under transmission electron microscope.Discussion and Conclusion
We found that while the controls had a fairly uniform interior, the lumazine expressing cells did not (Figure 2). In the lumazine cells there were homogenous areas of grey as well as darker spots, whereas the controls contained a more uniform interior.
We hypothesized that the lumazine accumulated together, which caused the isolation of other cellular elements. If this is the case, the microcompartments should most likely be the homogenous areas of grey not seen in the controls, and can’t be distinguished by contrast staining because they are aggregated together.
To see if this is the case we will look at cells expressing BBa_K542008 under the spectral confocal (fluorescence) microscope. We hypothesize that if the microcompartments are segregated from other cellular elements then the fluorescent proteins will also be segregated and therefore we should see areas within the cell with brighter fluorescence.Fluorescence Microscopy
E. coli DH5α cells containing BBa_K542008 were viewed with fluorescence microscopy. This was done in order to determine if the distribution of fluorescent proteins within the cells was similar to the homogenous grey regions seen in the cells under TEM.
Materials and Methods
E. coli DH5α cells containing the BBa_K542008 construct were grown overnight at 37°C. They were spun down at 10,000 rpm and then resuspended in 1X phosphate buffer saline (PBS). The solution was centrifuged at 10,000 rpm, decanted and resuspended in 4% paraformaldehyde (PFA) overnight. This solution was then centrifuged at 10000 rpm and cells were resuspended in glycerol. The glycerol-cell mixtures were then placed on slides, coverslipped and sealed.
The slides were viewed with an Olympus FV1000 spectral confocal at 60X magnification. Pictures were taken using the ECFP and EYFP filters.Results
Figure 4. E. Coli DH5a cells containing the BBa_K542008 construct as viewed under spectral confocal microscope with 60X magnification. The left picture corresponds to the ECFP filter and the right picture corresponds to the EYFP filter.Conclusion
Figure 4 shows that the fluorescent proteins were expressed in the cells. It is also evident that the fluorescence was segmented in a way that looks similar to that found in the homogenous grey areas in the TEM micrographs of the cells.
References
(1) Seebeck, F., Woycechowsky, K., Zhuang, W., Rabe, J., and Hilvert, D. (2006). A simple tagging system for protein encapsulation. Journal of the American Chemical Society. 128: 4516-4517.
Part: BBa_K542010 - Enhanced Lumazine Synthase (ELS)
Datasheet for Part BBa_K542010 in E. coli strain BL21 (DE3).
Introduction
Lumazine synthase (LS) from Aquifex aeolicus forms icosahedral microcompartment (MC) assemblies of 60 or 180 monomeric units that self assemble and are capable of isolating proteins from their local environment. As shown in previously published work (1), the LS protein has been mutated so that the interior of the MC is negatively charged; the UL 2009 iGEM team has submitted the mutated LS gene to the parts registry (BBa_K249002). A negatively charged interior allows for preferential compartmentalization of positively charged molecules, which can easily be engineered through the addition of a poly-arginine tag to a target protein (1). The size of the cavity of the Lumazine Synthase microcompartment was enlarged and the negative charge was increased via directed evolution (2). The compartment formed by this polypeptide has a larger cavity and a larger net negative charge, thus allowing a larger loading capacity into the cavity of the compartment.
The part was synthesized by Bio Basic Inc. into the pET28a plasmid vector (which allows for expression of ELS with an N-terminal His-tag). The Enhanced Lumazine Synthase (ELS) was moved into the pSB1C3 vector by the Lethbridge 2011 team.Characterization
Overexpression
The His-tagged ELS in pET28a was transformed into E. coli BL21 (DE3) and overexpressed.
Materials and Methods
4 X 500 mL of LB media in 2 L flasks were inoculated with E. coli BL21 (DE3) cells expressing ELS. Cells were grown at 37°C with shaking until the culture reached an OD600 of 0.683. The cultures were then induced with 10 mM IPTG. Culture samples containing equal amounts of cells were taken before induction and 30 min, 1 hour, 2 hours, and 3 hours post induction for analysis by SDS-PAGE. At 5 hours post induction the cells were harvested by centrifugation at 5000 x g for 5 min. The cell lysate was resuspended in 42 mL of buffer containing 50 mM Tris pH 8.0, 60 mM NH4CL, 7 mM β-mercaptoethanol, 1 mM PMSF, 7 mM MgCl2, 300 mM KCl, 10 mM imidazole, and 15% glycerol. Afterwards, 0.05 g of crystallized lysozyme was added to the cell suspension and incubated on ice for 1 hour. The lysate was then centrifuged for 70 minutes at 30000 x g to obtain the S30 fraction.
Results
Figure 1. 12.5% SDS-PAGE of His-tagged ELS Overexpressed in E. coli BL21 (DE3). Samples were taken at 1 hour increments after IPTG induction. Lane 1: zero hours; Lane 2: 1 hour; Lane 3: 2 hours; Lane 4: 3 hours. The expected molecular weight of enhanced lumazine synthase is 18.5 kD.Conclusion
Figure 1 clearly displays an increase in expression of a protein smaller than 25.0 kD, which is approximately the expected size of enhanced lumazine synthase monomers (18.5 kD).
Protein Purification
Enhanced lumazine synthase microcompartments were purified using size exclusion chromatography (SEC) to determine if the purified ELS formed a homogenous mixture of compartments, heterogenous mixture of microcompartments, or remained in monomer form.
Materials and Methods
In order to obtain a sample of ELS that was near homogeneity for characterization with SEC, the ELS sample was first purified by immobilized metal-ion affinity chromatography (IMAC). Ni2+-Sepharose beads supplied by Sigma Aldrich were pre-swelled in buffer containing 20% ethanol and 1mM NaCl. 3.75 mL of the Ni2+-Sepharose slurry was added to a 50 mL falcon tube. The beads were centrifuged at 500 x g for 2 min and the ethanol-containing buffer decanted. The beads were washed with 3 bed volumes of sterile water. The column was then washed in 6 bed volumes of Buffer A containing 50 mM Tris pH 8.0, 60 mM NH4CL, 7 mM β-mercaptoethanol, 1 mM PMSF, 7 mM MgCl2, 300 mM KCl, 10 mM imidazole, and 15% glycerol. S30 cell extract was applied to the column and mixed gently. The resin-S30 extract suspension was incubated on ice for 1 hour. The mixture was centrifuged at 500 x g for 2 min and the supernatant decanted. The supernatant was kept for further analysis by SDS-PAGE. The resin was washed 3 times with a full falcon tube volume (50 mL) of Buffer A. The mixture was centrifuged at 500 x g for 2 min and the supernatant decanted. The supernatant was kept for further analysis by SDS-PAGE. The column was washed 4 times with a full falcon tube volume (50 mL) of Buffer B (Buffer A plus 20 mM imidazole). The mixture was centrifuged at 500 x g for 2 minutes and the supernatant decanted. The supernatant was kept for further analysis by SDS-PAGE. The protein was eluted 9 times using 90% bed volume of Buffer E (Buffer A plus 250 mM imidazole). After each wash the sample was kept for analysis by SDS-PAGE.
Fractions 1 – 9 were pooled and concentrated using a Vivaspin Column with a 10 kDa molecular weight cut-off.
Concentrated protein samples from the Ni2+-Sepharose column were applied to a Sephacryl S400 size exclusion chromatography column at a flow rate of 0.4 mL/minute. 150 mL of SEC Buffer (50 mM sodium phosphate, 5 mM EDTA, 200 mM NaCl, pH 8.0, with 20% glycerol) was pumped through the column. The fractions eluted were collected and the absorbance was measured at 280 nm (see Fig 3 and 4).Results
Figure 2. SDS-PAGE (15%) of ELS purification by IMAC. Lane 1: protein marker. Lanes 2-10: Elution Fractions 1 – 9.
Figure 3. (A) Concentrated protein sample from the Ni2+-Sepharose column applied to a Sephacryl S400 size exclusion chromatography column at a flow rate of 0.4 mL/minute. The absorbance of the eluting solution was measured at 280nm. (B) Magnified view of chromatogram of the two peaks seen in (A).Conclusion
Figure 2 shows that with each wash proteins of a similar size were eluted off the resin. These proteins run at the expected molecular weight of enhanced lumazine synthase monomers (18.5 kD).
The chromatogram (Figure 3) from the chromatography experiment displays two regions of increased absorbance at 280 nm, between 82 and 135 mL of buffer eluted. These fractions were pooled, concentrated, and analyzed using transmission electron microscopy.Transmission Electron Microscopy
Purified samples of enhanced lumazine synthase from size exclusion chromatography (SEC) were characterized using transmission electron microscopy. We viewed samples of each of the two peaks from the SEC; however only one sample contained microcompartments.
Materials and Methods
Purified Samples in solution were placed on a carbon grid and negatively stained using uranyl acetate. Carbon grids containing the samples were then viewed with a Hitachi H-7500 Transmission Electron Microscope.
Results
Figure 4. Transmission electron microscopy of Enhanced Lumazine Synthase microcompartments. Microcompartments of approximately 40 nm can be seen at x 20K magnification.
Figure 5. Transmission electron microscopy of Enhanced Lumazine Synthase microcompartments. Microcompartments of approximately 40 nm can be seen at x 100K magnification.Conclusion
As expected, TEM micrographs show polyhedral particles of approximately 40 nm, corresponding to the size described by Wörsdörfer et al. (2011). This confirms that E. coli is capable of producing enhanced lumazine synthase.
References
(1) Viadiu H, Aggarwal AK (2000). Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol. Cell 5 (5): 889-895.
(2) iGEM 2007. (2011, May 10). BerkiGEM2007Present5. Retrieved from http://parts.mit.edu/igem07/index.php/BerkiGEM2007Present5.Part: BBa_I716462 – BamHI
Datasheet for Part BBa_I716462 in E. coli strain DH5alpha.
Introduction
Part I716462 was previously characterized by the UC Berkeley iGEM team in 2007. The purpose of this part was to prevent unwanted cell proliferation by engineering a genetic self-destruct mechanism into bacteria that upon induction would express a genetic material-degrading toxin, the endonuclease BamHI that would kill cells yet preserve their physical integrity. BamHI has a restriction cut site of G'GATCC, generating sticky ends (1). The restriction endonuclease was placed under the control of the arabinose- inducible promoter, pBAD. In 2007, the UC Berkeley iGEM team was able to show that cells lost their ability to reproduce but did not lyse upon induction with arabinose (2). The University of Lethbridge 2011 iGEM team wished to further characterize this part to degrade the genomic DNA of E. coli in order to have a functional chassis for our tailings pond clean-up kit without the risk of DNA contamination. In this way, tight control and regulation over our engineered bacterial cells is possible.
Characterization
Cell Survival
Materials and Methods
The BamHI construct was overexpressed in E. coli DH5α cells. Cultures were induced with arabinose at an OD600 of 0.6 and grown overnight. Samples of the induced culture and an uninduced culture were plated on LB agar plates to monitor growth of colony forming units.
Results
Figure 1. Growth curves of E. coli DH5α cells transformed with BamHI construct. Blue line represents cells induced at an OD600 of 0.60. Red line represents uninduced cells.
Figure 2. E.coliDH5α cells containing the BamHI construct from overnight cultures. The samples were plated on LB agar plates and the number of colony forming units (CFU) was counted. Left: culture induced for BamHI expression with arabinose, right: uninduced culture.Analysis of Genomic DNA
Materials and Methods
E. coli DH5α cells were grown and induced to overexpress one of three samples: BamHI construct induced with arabinose (10 µM) at an OD600 of 0.6, BamHI construct uninduced, non-BamHI construct induced with arabinose (10 µM) at OD600 of 0.6. Cells were grown in LB media with 100 ng/mL ampicillin.
Qiagen Blood and Tissue Kit was used to isolate the genomic DNA of gram-negative bacteria. A 0.8% agarose gel was ran at 150 V for 90 minutes to separate the genomic DNA of E. coli DH5α cells collected at different time points (pre-induced and 30 min, 1 hr, 2 hr, and 3 hr post induction) for all three samples.Results
Figure 3. Isolation of genomic DNA from E. coli DH5α cells. From left to right: 1Kb + ladder, 3hr, 2 hr, 1 hr, and 30 min after induction with arabinose and pre-induction for cells containing BamHI gene construct, uninduced control, and cells not containing the BamH1 gene construct.Conclusion
Upon induction with arabinose, E. coli DH5α cells containing the BamHI construct did not reproduce as the overexpression of the BamHI endonuclease digested the genomic DNA of the host cell. This is why in Figure 2 there are only 4 colonies seen as opposed to the control which has an uncountable amount of colonies. In Figure 1, the optical density of the BamHI expressing cells reaches a plateau; however, the same sample which has not been induced kept increasing, which indicates that the expression of BamHI is affecting normal growth of the cells.
Lastly, the genomic DNA of the E.coli cells was isolated. Upon overexpression of BamHI, there are still large segments of DNA at the pre-induced time point; however, as the post-induction time increases, there is an increase in digestion fragments.References
(1) Viadiu H, Aggarwal AK (2000). Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol. Cell 5 (5): 889-895.
(2) iGEM 2007. (2011, May 10). BerkiGEM2007Present5. Retrieved from http://parts.mit.edu/igem07/index.php/BerkiGEM2007Present5.
 "
"



























