 "
"
Team:Washington/Alkanes/Future/DecarbDesign
From 2011.igem.org
(→Methods) |
(→Methods) |
||
| Line 22: | Line 22: | ||
When OD600 reaches the value of 0.8, induce the culture with 50ul of 1M IPTG (final conc. 1mM) then shake the flasks at 37°C for ~3 hours | When OD600 reaches the value of 0.8, induce the culture with 50ul of 1M IPTG (final conc. 1mM) then shake the flasks at 37°C for ~3 hours | ||
Transfer to centrifuge tubes (50mL) and spin down (for how long?). Discard the liquid and put the tubes of concentrated cells (white dust at the bottom of the tube) into freezer for storage | Transfer to centrifuge tubes (50mL) and spin down (for how long?). Discard the liquid and put the tubes of concentrated cells (white dust at the bottom of the tube) into freezer for storage | ||
| + | |||
| + | Prepare stocks for protease inhibitor, DNAse, and lysozyme. | ||
| + | Protease inhibitor = 25x stock ; one tablet into 2ml of buffer | ||
| + | DNAses = 10mg/ml 100x stock (0.1mg/mL final) | ||
| + | Lysozyme = 10mg/ml 10x stock (1mg/mL final) | ||
| + | Resuspend 50ml cultures in 3ml of this mixture: 2.55ml sodium phosphate buffer, 120ul protease inhibitor, 30ul DNAse (0.1mg/ml final), and 300ul lysozyme (1mg/ml final), and XX uL 10% triton (1% final). Divide this solution into 3 effendorf tubes (each effendorf should contain 1ml of cell). | ||
| + | Prepare cell lysing buffers: | ||
| + | Sonicate --use micro tip, 60%, 30 on 30 off | ||
| + | Bugbuster --Use 10x bugbuster, follow protocol | ||
| + | Triton X-100 --Use 1% triton x100 which is equal to 10ml of 10% triton in 90ml of water. | ||
| + | Add bugbust/triton to appropriate effendorf tube and mix the tube at room temperature for 30 min. Then, centrifuge the tubes at max. speed for 15min (or until the liquid is very clear and not cloudy) | ||
| + | Transfer the liquid on top to new labeled effendorf tubes. | ||
| + | Place Target polyspring inserts into the Target DP glass vials (prepare 18 of this) | ||
| + | Set up 20 reactions in Target DP vials | ||
| + | S/B/T (3 different methods to lyse the cell) - 1/10 (2 different amount of cell lysing buffer) | ||
| + | ANF (6 rxns) AN (6 rxns) A (3 rxns) - This is negative control Alkane (4 rxns)- This is positive control | ||
| + | Cell1 10ul | ||
| + | Aldehyde 10ul | ||
| + | NADPH 28ul | ||
| + | Fere 1ul | ||
| + | Fere red 1ul Cell2 10ul | ||
| + | Aldehyde 10ul | ||
| + | NADPH 28ul | ||
| + | Buff 2ul Cell 10ul | ||
| + | Aldehyde 10ul | ||
| + | Buff. 30ul 100ul | ||
| + | 10ul | ||
| + | 1ul | ||
| + | 0.1ul | ||
| + | |||
| + | |||
| + | |||
| + | ----Make master mix in buffer of cofactors and aldehyde to add | ||
| + | 40 uL master mix, 10/1 uL cell lysate (all 50 uL rxns) | ||
| + | ***Resuspend lyophilized enzyme in 50% glycerol and phos buffer, into cold block, 100x ? 50X | ||
| + | 2.2 mg/mL stock ferredoxin (~50x) | ||
| + | Make enzyme up to 50x as well (fere red) 0.4ml of ferredoxin | ||
| + | |||
| + | Have powder NADPH, dissolve in buffer, make 2 mM, add 38 uL of this to master mix for ~1.5 mM (Make 20 mM NADPH initial) | ||
| + | From paper: | ||
| + | “To test the aldehyde decarbonylase (ADC) for activity, the following enzyme assays were set‐up: 200 μl reactions contained 100 mM sodium phosphate buffer at pH 7.2 with the following components at their respective final concentrations: 30 μM of purified ADC, 200 μM octadecanal, 50 μg/ml spinach ferredoxin (Sigma), 0.05 units/ml spinach ferredoxin reductase (Sigma), and 1 mM NADPH (Sigma). Negative controls included the above reaction without ADC, without octadecanal, or without spinach ferredoxin, ferredoxin reductase and NADPH. Each reaction was incubated at 37°C for 2 hours before being extracted with 100 μl ethyl acetate.” | ||
| + | ----We do 500 uM octadecanal | ||
*Protein Purification Assay | *Protein Purification Assay | ||
Revision as of 17:07, 12 September 2011
Contents |
Background
The objective of this subproject was to modify the ADC protein so that we can synthesize a wider variety of alkanes; in other words, mutate the protein so that it works better on a range of saturated fatty aldehydes. The protein was originally surmised to work best on C18 aldehydes, as the crystal structure derived Pymol model of the protein revealed a C18 carboxylic acid bounded to the metal center (though the vector with AAR and ADC proved to work best on C16 aldehydes while less on C14 and C18 aldehydes). Consequently, we decided to strive to modify ADC to work on shorter chain aldehydes, specifically tetradecanal. The substrate on the original Pymol file was modified to model C14 aldehyde, and then the file was converted to a Fold-it puzzle for human interaction. We decided to avoid changing amino acids near the active site, which binds to the aldehyde group, as we wanted to maintain the basic chemistry, decarbonylation (or perhaps deformylation). There was also the issue of the Fold-it modeling 122-Tyrosine hydrogen bonded to the substrate and another side chain, and according to Fold-it's notifications, serine, threonine, and tyrosine cannot "have more than 1 donor and 2 acceptors." As the amino acid we will change will be around the alkyl chain, there are no hydrogen bonds to be made(hydrogen bonds show up on the interface). If we are to make changes so that the protein binds more favorably to C14 aldehydes, we must improve dispersion intermolecular interactions by increasing the surface interaction between protein and substrate. Comparison with the original Pymol file showed that the removal of the four carbon atoms created a spatial “void area.” Targeting the void area with minimal interference or clashes between atoms, the most promising mutation sites are on adjacent sections of two helices positioned opposite of the substrate’s carbon end. The adjacent sections take up amino acids 21 to 25 and 67 to 71, the former of which, being closer, shows promise as a site to mutate to fill in the void area and avoid interferences.
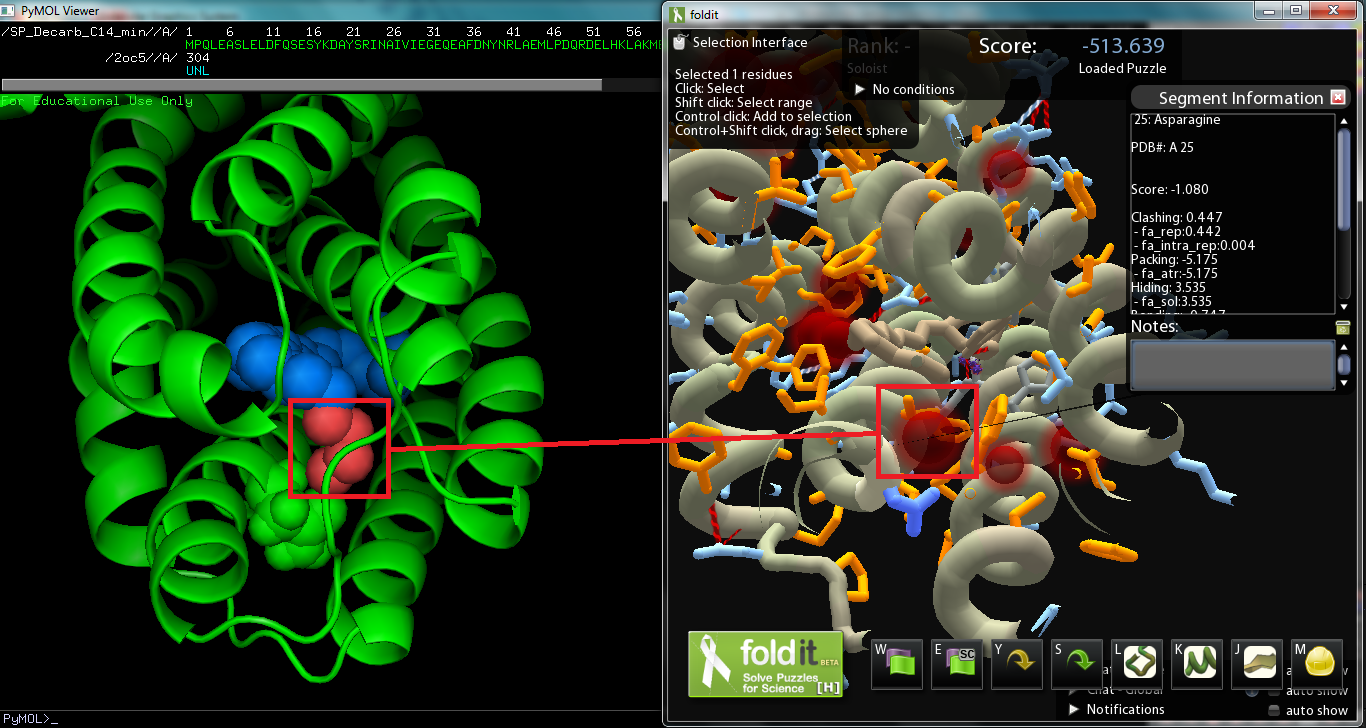

Methods
- In Vitro Assay (Cell Lysis Assay)
Obtain a15mL Falcon Tube. Add 2mL of TB and add 2ul of 1000x Kanamycin into the Falcon Tube. Obtain a tiny colony from the agar plate with the cells. If you are obtaining colony from a glycerol stock, swirl the surface of the glycerol stock and place it into the Falcon tube. Put the tubes in the shaker overnight at 37 degree. Get two 250ml flasks and wash them cleanly and seal the top of each flask with foil. Autoclave two 250ml flasks. Before you start autoclaving the machine, make sure that the door of the machine is completely shut. Also, at the end of autoclaving, make sure that the machine has been fully depressurized and cooled down. Add 50ml of LB media into each of the two autoclaved 250ml flasks. Add 500ul of the overnight culture into the each flask. Add 50ul of kanamycin into the each flask. Place the two flasks in the 37°C shaker and begin growing cells. When OD600 reaches the value of 0.8, induce the culture with 50ul of 1M IPTG (final conc. 1mM) then shake the flasks at 37°C for ~3 hours Transfer to centrifuge tubes (50mL) and spin down (for how long?). Discard the liquid and put the tubes of concentrated cells (white dust at the bottom of the tube) into freezer for storage
Prepare stocks for protease inhibitor, DNAse, and lysozyme. Protease inhibitor = 25x stock ; one tablet into 2ml of buffer DNAses = 10mg/ml 100x stock (0.1mg/mL final) Lysozyme = 10mg/ml 10x stock (1mg/mL final) Resuspend 50ml cultures in 3ml of this mixture: 2.55ml sodium phosphate buffer, 120ul protease inhibitor, 30ul DNAse (0.1mg/ml final), and 300ul lysozyme (1mg/ml final), and XX uL 10% triton (1% final). Divide this solution into 3 effendorf tubes (each effendorf should contain 1ml of cell). Prepare cell lysing buffers: Sonicate --use micro tip, 60%, 30 on 30 off Bugbuster --Use 10x bugbuster, follow protocol Triton X-100 --Use 1% triton x100 which is equal to 10ml of 10% triton in 90ml of water. Add bugbust/triton to appropriate effendorf tube and mix the tube at room temperature for 30 min. Then, centrifuge the tubes at max. speed for 15min (or until the liquid is very clear and not cloudy) Transfer the liquid on top to new labeled effendorf tubes. Place Target polyspring inserts into the Target DP glass vials (prepare 18 of this) Set up 20 reactions in Target DP vials S/B/T (3 different methods to lyse the cell) - 1/10 (2 different amount of cell lysing buffer) ANF (6 rxns) AN (6 rxns) A (3 rxns) - This is negative control Alkane (4 rxns)- This is positive control Cell1 10ul Aldehyde 10ul NADPH 28ul Fere 1ul Fere red 1ul Cell2 10ul Aldehyde 10ul NADPH 28ul Buff 2ul Cell 10ul Aldehyde 10ul Buff. 30ul 100ul 10ul 1ul 0.1ul
Make master mix in buffer of cofactors and aldehyde to add
40 uL master mix, 10/1 uL cell lysate (all 50 uL rxns)
- Resuspend lyophilized enzyme in 50% glycerol and phos buffer, into cold block, 100x ? 50X
2.2 mg/mL stock ferredoxin (~50x) Make enzyme up to 50x as well (fere red) 0.4ml of ferredoxin
Have powder NADPH, dissolve in buffer, make 2 mM, add 38 uL of this to master mix for ~1.5 mM (Make 20 mM NADPH initial) From paper: “To test the aldehyde decarbonylase (ADC) for activity, the following enzyme assays were set‐up: 200 μl reactions contained 100 mM sodium phosphate buffer at pH 7.2 with the following components at their respective final concentrations: 30 μM of purified ADC, 200 μM octadecanal, 50 μg/ml spinach ferredoxin (Sigma), 0.05 units/ml spinach ferredoxin reductase (Sigma), and 1 mM NADPH (Sigma). Negative controls included the above reaction without ADC, without octadecanal, or without spinach ferredoxin, ferredoxin reductase and NADPH. Each reaction was incubated at 37°C for 2 hours before being extracted with 100 μl ethyl acetate.”
We do 500 uM octadecanal
- Protein Purification Assay
- In Vivo Assay
Current Status
- Discuss mutations made and construct submitted
- Show protein gel of expression
- Discuss in vitro assay and how it hasn't worked yet (even for WT)
- Discuss plan for future testing
Parts Submitted
- Plasmids with mutations (ADC_AB4, 1C2, 7A5, 8BE, 9A1, 10B5)