 "
"
Team:BU Wellesley Software/Wet Lab
From 2011.igem.org
| Line 147: | Line 147: | ||
The composite parts made by the wet lab have been verified using restriction mapping, where each part is digested and the resulting fragments are analyzed based on their band sizes. For a more detailed documentation of the wet lab works, please look at the individual notebook.<br> | The composite parts made by the wet lab have been verified using restriction mapping, where each part is digested and the resulting fragments are analyzed based on their band sizes. For a more detailed documentation of the wet lab works, please look at the individual notebook.<br> | ||
<br> | <br> | ||
| - | <img | + | <img src="https://2011.igem.org/File:Arabinosediagram.jpg"> |
Despite our plan to create a reconfigurable plasmid by the end of summer, we encountered some problems that prevented us from achieving that. In the first few days, we had an extremely low DNA quantification values for the plasmid. While the actual number of plasmid in the liquid culture may vary depending on whether the plasmid is a high or low copy, no DNA could be visualized in gel electrophoresis. In order to confirm if the correct bacteria grew in the liquid culture, a small sample was taken for gram staining and visualization under the microscope. It appeared that there were lots of other bacteria that got amplified, probably because the antibiotic selection did not work or our lab equipment got contaminated. So we autoclaved the pipet tips and prepared a new batch of antibiotic-equipped LB agar plates. Then, we had a problem with ligation where no colonies appeared on the transformed plates. We tried tinkering with the ligation protocols by using different insert to vector ratio, ethanol precipitation, and various enzymes. After several unsuccessful troubleshooting approaches, we tried pelleting the cell and using a commercial line of competent cell. Fortunately, the second method works and lots of composite parts were made afterwards. The final problem involved the failure to induce the pBad promoter. After adding various concentration of arabinose and using different protocols, it turned out that the commercial cell lines that we have been using did not contain the AraC gene. Since the AraC gene has to exist and interact with arabinose to activate the pBad promoter, we then looked up the part that contains AraC gene and added it to the existing part. While no fluorescence was been observed under UV, the analysis of those samples with the FACS machine has produced a time series data that showed significant rise in fluorescence intensity after the induction with arabinose.<br> | Despite our plan to create a reconfigurable plasmid by the end of summer, we encountered some problems that prevented us from achieving that. In the first few days, we had an extremely low DNA quantification values for the plasmid. While the actual number of plasmid in the liquid culture may vary depending on whether the plasmid is a high or low copy, no DNA could be visualized in gel electrophoresis. In order to confirm if the correct bacteria grew in the liquid culture, a small sample was taken for gram staining and visualization under the microscope. It appeared that there were lots of other bacteria that got amplified, probably because the antibiotic selection did not work or our lab equipment got contaminated. So we autoclaved the pipet tips and prepared a new batch of antibiotic-equipped LB agar plates. Then, we had a problem with ligation where no colonies appeared on the transformed plates. We tried tinkering with the ligation protocols by using different insert to vector ratio, ethanol precipitation, and various enzymes. After several unsuccessful troubleshooting approaches, we tried pelleting the cell and using a commercial line of competent cell. Fortunately, the second method works and lots of composite parts were made afterwards. The final problem involved the failure to induce the pBad promoter. After adding various concentration of arabinose and using different protocols, it turned out that the commercial cell lines that we have been using did not contain the AraC gene. Since the AraC gene has to exist and interact with arabinose to activate the pBad promoter, we then looked up the part that contains AraC gene and added it to the existing part. While no fluorescence was been observed under UV, the analysis of those samples with the FACS machine has produced a time series data that showed significant rise in fluorescence intensity after the induction with arabinose.<br> | ||
<br> | <br> | ||
Revision as of 19:09, 10 September 2011

Wet Lab
Background
While some bacterial infections are seen as common place, others remain elusive to cure.
Tuberculosis has currently infected a third of the world’s population, and 1.7 million people
died from it in 2009. As shown, it is a worldwide problem that is prevalent in third world
countries.
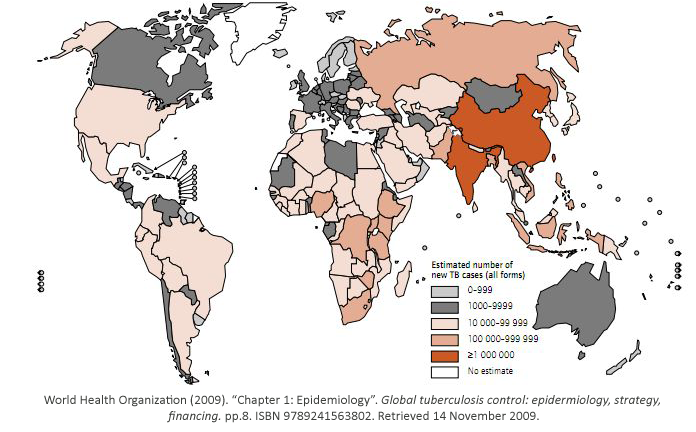
Tuberculosis exists in two stages. In its latent stage, the immune system has it under
control and prevents the bacteria from reproducing. When it switches into its active stage as
a person's immune system becomes compromised, it begins to multiply and the person begins
showingsymptoms of the disease.  The genetic mechanisms that trigger the change between the two
forms are not very well understood. The genetic network of tuberculosis is complicated and
studying the interactions between genes can be time consuming, requiring the construction of
many plasmids to study only one small part of the network. To look for new, faster ways of
studying this problem we turned to synthetic biology. Instead of creating many vectors
to study the interactions of genes, we aimed to create a plasmid with a designed circuit that
would control the transcription of several tuberculosis genes using invertases. We chose to
use non-pathogenic mycobacterium tuberculosis genes and transcription factors within non-
pathogenic E.Coli for safety reasons.
The genetic mechanisms that trigger the change between the two
forms are not very well understood. The genetic network of tuberculosis is complicated and
studying the interactions between genes can be time consuming, requiring the construction of
many plasmids to study only one small part of the network. To look for new, faster ways of
studying this problem we turned to synthetic biology. Instead of creating many vectors
to study the interactions of genes, we aimed to create a plasmid with a designed circuit that
would control the transcription of several tuberculosis genes using invertases. We chose to
use non-pathogenic mycobacterium tuberculosis genes and transcription factors within non-
pathogenic E.Coli for safety reasons.
Invertases are recombinases that will bind to recognition sites. Once bound, the DNA between two sites will invert and flip horizontally. This effectively acts as an on/off switch for transcription of that region of DNA as it is not available for DNA polymerase to bind to. In terms of circuit design, it will act as a not gate. Using them within a plasmid with several tuberculosis genes would allow us to check for regulatory measures such as negative feedback, etc.

An example of a construct built around the invertase architecture.
In addition to the novel cellular architecture, we also used a variety of software tools created by our computational teams (Boston University, Wellesley):
- G-nome Surfer Pro
G-nome Surfer Pro is an integrated environment that allows for the viewing of prokaryotic genomic data and literature associated with the genome. As it is built on a Microsoft Surface, it encourages collaboration. We used the Primer Genie to design the primers for the tuberculosis genes and transcription factors. - Trumpet
Trumpet consists of a library of genes and promoters, which can be configured into any desired permutation or combination by treating the DNA with recombinases, which allows us to rewire the genes and switches and study all their combinations. It was used by the wetlab to help us map out where the invertases should go in our plasmids in order to correctly turn on and off the segments of DNA we want to study. - PuppetShow
This suite includes a high level programming language for specifying biological protocols commonly used in the laboratory, which are then executed by a liquid-handling robot with minimal user intervention. We used this in conjunction with the robot (described below) to create samples that were simultaneously created manually to compare the results. - Clotho
Clotho is used to mange, create and store new biological building blocks in community based repositories. It includes a suite of tools that include PuppetShow and Trumpet, which were designed specifically for this project. Other tools that were used were:- Bull Trowel
- Spreadit Parts
- Spreadit Vectors
- Spreadit Features
- Optimus Primer-primer designer
- Feature Chomp-reads in APE and GENBANK files and takes the features found within the file to a feature database
- Batterboard- allowed us to electronically represent physical samples in the lab
Another way we looked into facilitating progress in studying genetic networks is through the use of automation:
- Robot
- QIA Cube
Results
[[File:PRET.jpg]]
The most important achievement in the wet lab is the creation of several genetic devices that can function as a reporter. Each of the part contains at least 1 of promoter, RBS, fluorescent gene, and terminator. The inclusion of these individual parts allows our devices to be fully characterized through the Fluorescence-Activated Cell Sorting (FACS) machine. Most of the devices have similar parts or construction method, which allow individual part to be characterized independently from the possible interaction with other parts. One concrete example for this concept is the creation of pTet+GFPc (Bba_R0040_E0240) and pCons+GFPc (Bba_R2000_E0240). Since the only difference between those 2 parts are only the promoter while everything else is virtually the same, it is possible to understand how each promoter influence a similar construct through the characterization data. The complete list of parts that we have made during the summer is: […]
The characterization data is […]
The composite parts made by the wet lab have been verified using restriction mapping, where each part is digested and the resulting fragments are analyzed based on their band sizes. For a more detailed documentation of the wet lab works, please look at the individual notebook.
 Despite our plan to create a reconfigurable plasmid by the end of summer, we encountered some problems that prevented us from achieving that. In the first few days, we had an extremely low DNA quantification values for the plasmid. While the actual number of plasmid in the liquid culture may vary depending on whether the plasmid is a high or low copy, no DNA could be visualized in gel electrophoresis. In order to confirm if the correct bacteria grew in the liquid culture, a small sample was taken for gram staining and visualization under the microscope. It appeared that there were lots of other bacteria that got amplified, probably because the antibiotic selection did not work or our lab equipment got contaminated. So we autoclaved the pipet tips and prepared a new batch of antibiotic-equipped LB agar plates. Then, we had a problem with ligation where no colonies appeared on the transformed plates. We tried tinkering with the ligation protocols by using different insert to vector ratio, ethanol precipitation, and various enzymes. After several unsuccessful troubleshooting approaches, we tried pelleting the cell and using a commercial line of competent cell. Fortunately, the second method works and lots of composite parts were made afterwards. The final problem involved the failure to induce the pBad promoter. After adding various concentration of arabinose and using different protocols, it turned out that the commercial cell lines that we have been using did not contain the AraC gene. Since the AraC gene has to exist and interact with arabinose to activate the pBad promoter, we then looked up the part that contains AraC gene and added it to the existing part. While no fluorescence was been observed under UV, the analysis of those samples with the FACS machine has produced a time series data that showed significant rise in fluorescence intensity after the induction with arabinose.
Despite our plan to create a reconfigurable plasmid by the end of summer, we encountered some problems that prevented us from achieving that. In the first few days, we had an extremely low DNA quantification values for the plasmid. While the actual number of plasmid in the liquid culture may vary depending on whether the plasmid is a high or low copy, no DNA could be visualized in gel electrophoresis. In order to confirm if the correct bacteria grew in the liquid culture, a small sample was taken for gram staining and visualization under the microscope. It appeared that there were lots of other bacteria that got amplified, probably because the antibiotic selection did not work or our lab equipment got contaminated. So we autoclaved the pipet tips and prepared a new batch of antibiotic-equipped LB agar plates. Then, we had a problem with ligation where no colonies appeared on the transformed plates. We tried tinkering with the ligation protocols by using different insert to vector ratio, ethanol precipitation, and various enzymes. After several unsuccessful troubleshooting approaches, we tried pelleting the cell and using a commercial line of competent cell. Fortunately, the second method works and lots of composite parts were made afterwards. The final problem involved the failure to induce the pBad promoter. After adding various concentration of arabinose and using different protocols, it turned out that the commercial cell lines that we have been using did not contain the AraC gene. Since the AraC gene has to exist and interact with arabinose to activate the pBad promoter, we then looked up the part that contains AraC gene and added it to the existing part. While no fluorescence was been observed under UV, the analysis of those samples with the FACS machine has produced a time series data that showed significant rise in fluorescence intensity after the induction with arabinose.
Because the main focus of our team is automation and software integration, this paragraph will talk about how several software tools are utilized. We used QIAcube to automate plasmid isolation and gel extraction protocols, allowing us to complete other tasks in the meantime. But the most important software utilized in our wet lab environment is Clotho. Clotho is a software suite that contains various synthetic biology applications that can assist us in constructing a genetic device. With Batterboard, we maintained an electronic database to keep track of our physical DNA samples. This is important because some protocols (i.e. restriction digest and ligation) have a quantitative nature: they require a calculation that involves DNA concentration value and/or plasmid size. Of course, some problems cannot be avoided while integrating the software tools into the wet lab as it requires a strong understanding of both molecular biology and computer programming. One important problem is [Clotho Experiences – Vanessa]
In addition to the software tools, another important goal from this project is the automation of molecular biology protocols. Using Puppet Show, we have developed a generalized laboratory protocol format that can be utilized both by the scientist and the EVO 10000 Liquid Handling Robot. [AUTOMATION – Evelyn]
We have conducted practice trials to try the Genome Surfer, an interactive tabletop interface for genetic research. This method allows the visualization of a hierarchical view from the chromosome, genes, up till the actual sequence. We had been trying out Primer Designer to help us finding a primer that fulfills the requirements. We have been learning about Trumpet to learn about the application of sorting algorithm in preparing reconfigurable plasmid architecture. But we have not implemented the last 2 methods in the laboratory work, since the previously mentioned problems left us with not enough time to actually producing even a simplified model of reconfigurable plasmid and characterizing it.
Approach
Lorem ipsum dolor sit amet, consectetur adipiscing elit. Quisque ut tincidunt quam. Praesent in sodales mi. In hac habitasse platea dictumst. Nunc magna massa, gravida in euismod nec, rutrum sed dolor. Sed accumsan ullamcorper quam nec feugiat. Nam interdum volutpat nibh nec ultrices. Pellentesque habitant morbi tristique senectus et netus et malesuada fames ac turpis egestas. Donec eget nibh leo. Nullam euismod vulputate leo, eu egestas dolor volutpat at. Vivamus ac purus mi, vitae consequat enim.