From 2011.igem.org
(Difference between revisions)
|
|
| (5 intermediate revisions not shown) |
| Line 148: |
Line 148: |
| | **Placed colonies in 20 μL of water and used 1 μl for a 20 μL PCR reaction | | **Placed colonies in 20 μL of water and used 1 μl for a 20 μL PCR reaction |
| | **Then took 10 μL from 20 μL dilution and added to 100 μL of LB in 96 well plate and put in 30°C fridge, at 3:19 pm | | **Then took 10 μL from 20 μL dilution and added to 100 μL of LB in 96 well plate and put in 30°C fridge, at 3:19 pm |
| - | **Same protocol as [[Protocols#PCR|KAPA PCR]] with the following changes made | + | **Same protocol as [https://2011.igem.org/Team:Harvard/Protocols#PCR KAPA PCR] with the following changes made |
| | ***Step 3, gradient for annealing, 55°C - 77°C, for a range of 55,57,59,61 and 63°C for the two rows of 5 PCR tubes each (placed leftmost) | | ***Step 3, gradient for annealing, 55°C - 77°C, for a range of 55,57,59,61 and 63°C for the two rows of 5 PCR tubes each (placed leftmost) |
| | ***Step 4, Extension, 1 min | | ***Step 4, Extension, 1 min |
| Line 154: |
Line 154: |
| | **Name: TOLCGRAD, PCR5 machine.</div> | | **Name: TOLCGRAD, PCR5 machine.</div> |
| | <div id="719" style="display:none"> | | <div id="719" style="display:none"> |
| | + | |
| | ==July 19th== | | ==July 19th== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 162: |
Line 163: |
| | | | |
| | ====Minipreps of colonies and controls==== | | ====Minipreps of colonies and controls==== |
| - | Our minipreps from [[#Colony minipreps|Saturday]] had low yields, especially for culture 85. We suspect this is why the PCR [[#Gel analysis of miniprep junction PCR|on Sunday]] did not work for this sample. Unfortunately, 85.3 and 85.4 did not grow even after being left in the shaker overnight. These were two cultures we made by picking new colonies from plate 85. This may have occurred because we did not actually pick the colony, or the colony was a cheater. Thus, we performed minipreps on 80.1, 80.2, 81.1, 81.4 (the controls, chosen for their wide range of concentrations after our first miniprep), and 85.1 and 85.2. | + | Our minipreps from Saturday had low yields, especially for culture 85. We suspect this is why the PCR [[#Gel analysis of miniprep junction PCR on Sunday did not work for this sample. Unfortunately, 85.3 and 85.4 did not grow even after being left in the shaker overnight. These were two cultures we made by picking new colonies from plate 85. This may have occurred because we did not actually pick the colony, or the colony was a cheater. Thus, we performed minipreps on 80.1, 80.2, 81.1, 81.4 (the controls, chosen for their wide range of concentrations after our first miniprep), and 85.1 and 85.2. |
| | | | |
| | In order to figure our why our minipreps were resulting in unexpectedly low yields and increase our yields: | | In order to figure our why our minipreps were resulting in unexpectedly low yields and increase our yields: |
| Line 179: |
Line 180: |
| | | | |
| | ====PCR of miniprep products==== | | ====PCR of miniprep products==== |
| - | We then ran a PCR for the cross-junction on all 6 (80.1, 80.2, 81.1, 81.4, 85.1, 85.2) samples, using [[#PCR of expression plasmid cross-junction|our usual cross junction protocol]]. | + | We then ran a PCR for the cross-junction on all 6 (80.1, 80.2, 81.1, 81.4, 85.1, 85.2) samples, using our usual cross junction protocol. |
| | | | |
| | {| | | {| |
| Line 191: |
Line 192: |
| | | | |
| | ====Designing ultramers for Zif268, OZ052, and OZ123==== | | ====Designing ultramers for Zif268, OZ052, and OZ123==== |
| - | In preparation for designing our 96-well practice plate, we designed ultramers for our three positive controls Zif268, OZ052, and OZ123. These ultramers contain the Type II binding sites as well as the F2/F3 fingers. Since the [[Lab_Notebook#Design_of_Plate_Practice_Sequences|practice plate]] will contain oligos that encode for F1, we can use these three positive controls to practice and ensure that we secure the technique of using the Type II binding sites to swap in F1 into our expression plasmids. The ultramers have not yet been ordered. | + | In preparation for designing our 96-well practice plate, we designed ultramers for our three positive controls Zif268, OZ052, and OZ123. These ultramers contain the Type II binding sites as well as the F2/F3 fingers. Since the practice plate will contain oligos that encode for F1, we can use these three positive controls to practice and ensure that we secure the technique of using the Type II binding sites to swap in F1 into our expression plasmids. The ultramers have not yet been ordered. |
| | | | |
| | ===Team TolC=== | | ===Team TolC=== |
| Line 308: |
Line 309: |
| | | | |
| | ====Construction of OZ052 and OZ123: Joining the Finger with Homology regions==== | | ====Construction of OZ052 and OZ123: Joining the Finger with Homology regions==== |
| - | Previously, we did a PCR to [[Lab_Notebook:_July#Overlap_PCR_of_OZ052_and_OZ123|reassemble the ultramers]] for OZ052 and OZ123, but they only coded strictly for F1/F2/F3 and did not have any overhang homology with any surrounding code. Thus, we performed PCR to add these homology overhangs so that we can integrate them with the omega subunit as well as the spec backbone to create a final expression plasmid. | + | Previously, we did a PCR to reassemble the ultramers for OZ052 and OZ123, but they only coded strictly for F1/F2/F3 and did not have any overhang homology with any surrounding code. Thus, we performed PCR to add these homology overhangs so that we can integrate them with the omega subunit as well as the spec backbone to create a final expression plasmid. |
| | | | |
| | The program we used was 55* annealing temperature, and a 15 second extension time. Gel results pictured below. | | The program we used was 55* annealing temperature, and a 15 second extension time. Gel results pictured below. |
| Line 365: |
Line 366: |
| | Our concentrations were better than they have been in the past; notably, after the addition of buffer P2, our solution actually turned a bright blue (indicating lysis of the cells), which we had not seen before. | | Our concentrations were better than they have been in the past; notably, after the addition of buffer P2, our solution actually turned a bright blue (indicating lysis of the cells), which we had not seen before. |
| | | | |
| - | We then ran a PCR on our miniprep product, to check for our desired insert. We used our [[#PCR of expression plasmid cross-junction|usual protocol]] for a PCR of the expression plasmid cross-junction. | + | We then ran a PCR on our miniprep product, to check for our desired insert. We used our usual protocol for a PCR of the expression plasmid cross-junction. |
| | | | |
| | {| | | {| |
| Line 374: |
Line 375: |
| | | | |
| | ====Sending out our expression colonies for sequencing==== | | ====Sending out our expression colonies for sequencing==== |
| - | We sent out the samples from [[#Expression Plasmid Sequencing Results|yesterday]] that had no more than 1 SNP for reverse sequencing. In addition, we also sent out 78 and 79 for forward and reverse sequencing. | + | We sent out the samples from yesterday that had no more than 1 SNP for reverse sequencing. In addition, we also sent out 78 and 79 for forward and reverse sequencing. |
| | *Reverse sequencing: 77.1, 77.3, 77.4, 78.1, 80.1, 80.2, 80.4, 81.1, 81.4, 82.1, 83.4, 84.1, 84.4, Zif268 | | *Reverse sequencing: 77.1, 77.3, 77.4, 78.1, 80.1, 80.2, 80.4, 81.1, 81.4, 82.1, 83.4, 84.1, 84.4, Zif268 |
| | *Forward and reverse sequencing: 781, 78.2, 78.3, 78.4, 79.1, 79.2, 79.3, 79.4 | | *Forward and reverse sequencing: 781, 78.2, 78.3, 78.4, 79.1, 79.2, 79.3, 79.4 |
| Line 389: |
Line 390: |
| | | | |
| | ====Gel Extraction and Isothermal Assembly of OZ fingers+homology==== | | ====Gel Extraction and Isothermal Assembly of OZ fingers+homology==== |
| - | We ran a gel extraction of our PCR product from [[#Construction of OZ052 and OZ123: Joining the Finger with Homology regions|yesterday]], so that we could use it for isothermal assembly later. Strangely, we got bands that were larger than our largest theoretical product. | + | We ran a gel extraction of our PCR product from yesterday, so that we could use it for isothermal assembly later. Strangely, we got bands that were larger than our largest theoretical product. |
| | | | |
| | {| | | {| |
| Line 422: |
Line 423: |
| | *Also replated the 7/20 lambda red culture to see if we can get any good cells (1.5mL).</div> | | *Also replated the 7/20 lambda red culture to see if we can get any good cells (1.5mL).</div> |
| | <div id="722" style="display:none"> | | <div id="722" style="display:none"> |
| | + | |
| | ==July 22nd== | | ==July 22nd== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 452: |
Line 454: |
| | | | |
| | | | |
| - | To this end, we redid the PCRs for everything in preparation for the resequencing, using the junction primers and [[#PCR of expression plasmid cross-junction|the previous protocol]]. The resulting gels can be observed below: | + | To this end, we redid the PCRs for everything in preparation for the resequencing, using the junction primers and the previous protocol. The resulting gels can be observed below: |
| | | | |
| | {| | | {| |
| Line 467: |
Line 469: |
| | | | |
| | ====Transformation for OZ plasmids==== | | ====Transformation for OZ plasmids==== |
| - | Today, we took the results of the isothermal assembly from yesterday and did chemical transformation to introduce the plasmids into the Top 10 ChemComp bacteria, using [[Protocols#Cultures|our protocol for chemical transformations]]. They were left to recover for approximately 1.5 hours. We plated 2 dilutions, 10 ul and 50 ul, and left them overnight at 37* in the incubator. | + | Today, we took the results of the isothermal assembly from yesterday and did chemical transformation to introduce the plasmids into the Top 10 ChemComp bacteria, using [https://2011.igem.org/Team:Harvard/Protocols#Cultures our protocol for chemical transformations]. They were left to recover for approximately 1.5 hours. We plated 2 dilutions, 10 ul and 50 ul, and left them overnight at 37* in the incubator. |
| | | | |
| | ===Team Wolfe=== | | ===Team Wolfe=== |
Latest revision as of 20:53, 4 August 2011
July 15th
Team Wolfe
Thio kan-ZFB-wp-hisura optimizing PCR results:
- The gel stab PCR produced a product of the right size, but the 2kb side product was still brighter. The 2-PCR protocol also still had substantial side products but at least they did not completely overshadow the real product. We will accordingly go ahead and do a gel extraction on the 2-PCR samples so that we can try lambda red with them next week.

Different attempts and thio kan-ZFB-hisura PCR 7/15/11
pSR01 growth phenotype results:
- Unfortunately the computer hooked up to the plate reader spontaneously restarted during the night to download updates, so we lost all the data. Just by looking at the plate, it still seems like IPTG is inhibiting growth. The ∆HisB∆pyrF∆rpoZ controls also did not grow, probably because the plate the colonies were taken from was too old, so we will restreak from the glycerol stock and set up the plate reader again this weekend.
Team ZF
PCR of electro comp expression plasmids
We performed a mini-prep of the cultures that we grew overnight of the electrocompetent cells. We then performed a PCR on the product of the mini-prep, checking for the 1.4 kb cross junction to confirm the presence of our omega+ultramers+spec plasmid. We used the same recipe as the PCR we performed on Tuesday.
 Description of gel below. |
No bands appeared for PCR peformed on a 1 to 10 dilution of the cultures. Miniprep product from colonies 78, 79, and 82 had the expected band at 1.4 kb, while 80 and 81 had a band that was slightly smaller than the the expected product size. We saw a similar band in our PCR of the isothermal assembly reaction product. We are unsure exactly what is causing this band. If either the omega subunit or ultramers were missing from the plasmid, a similar sized product would result since both are ~350 bp. We are curious if a plasmid can still be formed if one of these components were missing.
We checked the concentrations of our miniprep product to see if that would explain our problem.
ID Conc. (ng/ul) 260/280 1.4 kb band?
78 16.12 2.10 Y
79 41.64 1.64 Y
80 5.84 2.01 N
81 61.24 1.62 N
82 20.25 1.72 Y
We can't tell whether the concentration of the miniprep product contributed to the strange band we saw. 81 had the highest concentration but still didn't work. We decided to try plating 4 more colonies from 80 and 81, to see if any of them have the appropriate band.
We also decided to make final plates of cultures 78, 79, and 82, using 2 ul of culture and 50 ul of LB+spec. They were plated on new spec plates. We will incubate them at 37* overnight.
Checking the expression plasmid plates
All the plates had colonies on them, although there were still areas of unspecific growth, presumably because of uneven dilution of the spec into the LB plates.
We made cultures of all the chem comp cells (77, 83, 84, 85) and the electro comp cell that did not have the correct insertion (80, 81). We picked 4 colonies from each plate, resulting in a total of 24 cultures. We have left them in the 37* shaker overnight, and tomorrow plan on performing a miniprep and PCR to check for the cross-junction.
PCR to check for omega+ultramers
We performed a PCR on the five electro comp miniprep products, to find out what may be missing. We used the omega_F+pLacO (index: 73) as our forward primer, and each of the respective reverse ultramers as our reverse primers (index: 78-82). We had an annealing temperature of 58* and an extension time of 30 seconds, and the expected product size is 646 bp.
 Team ZF results in lanes 8-12 |
Strangely, we did not get results for sample 78 (FH bottom), even though we got a band there in a PCR earlier today that used the junction primers. We got positive results for samples 79 and 82, and everything else seems to check out with these two samples (bands present in the correct size with junction primers from both isothermal assembly and miniprep templates). However, we are still having problems with samples 80 and 81, which may be a problem with the colonies that we picked. For now, we will assume that the isothermal assembly worked, due to colonies growing on the spec plates for samples 80 and 81, and that we had simply picked bad colonies. We will find out tomorrow if other colonies from the same plate give us the same results.
July 16th
Team ZF
Colony minipreps
Minipreps were performed on all of the samples except for a few where nothing grew in the LB/spec media. These exceptions included 85.3, 85.4, 84.2, 81.2, and 81.3. Possible reasons for no growth for these samples may include: (1) The colony was somehow not a transformed colony and was a cheater (possibly picked from a spot where there was a smaller amount of spec in the unevenly distributed dish), and (2) the colony did not get picked up properly with the pipet tip.
The concentrations of the DNA after miniprep were somewhat low, and a full table of these concentrations and DNA purities can be found below:
PCR on minipreps to check cross junction
PCR was done on the miniprep products to check for the cross junction length, expected band size of ~1.4 kb. The protocol used was the same as the previous time. In addition to PCR-ing the miniprep products, a positive control was also included, which was the finished Zif268 + omega + spec backbone plasmid (this was the first successful junction PCR we obtained, and so it should work again).
Replating final samples for 78, 79, 82
Strangely, nothing grew on the plates that we seeded yesterday with 2ul of bacteria. We seeded plates again for samples 78, 79, and 82 with 15ul of transformed bacteria instead, and hopefully these should grow by tomorrow.
July 17th
Team ZF
Replating final samples, take 2
We realized that the plates we seeded yesterday (July 16) were seeded from the transformation bacteria tubes, which has a mix of bacteria that may/may not have taken up the plasmid. These plates therefore cannot serve as the final stock plates. We took the E. coli from our initial colony picking and put them in LB + spec to grow to saturation tomorrow, where we will replate again with those bacteria (for which the good PCR results should correlate).
Gel analysis of miniprep junction PCR
The miniprep junction PCR products from yesterday were ran on an agarose gel, with the results below:
 PCR Results, expected band at ~1.4 kb |
While all the bands are between 1000 and 1650 kb, there are slight variations in the size for which we are unable to account. We will send these out for sequencing, to try to figure out what is going on.
July 18th
Team ZF
Sending expression plasmids out for sequencing
We decided to send out the expression plasmids that had bands at approximately the correct size out for sequencing. We originally wanted to sequence the plasmids, but our miniprep concentrations were not high enough. Apparently, we should be getting high yields from minipreps, but thus far our yields have been around 50 ng/ul at the highest.
We decided to sequence the unpurified PCR products instead. In order to do this we had to re-do the PCRs for Zif268, FH bottom (78), CB top (79), Myc 981 (82). Below are our 1% e-gel results, verifying that we had appropriately sized bands:
 Gel of cross junction of Zif268, FH bottom, CB top, Myc 981 |
Our final samples sent out for sequencing are:
Index Sample ID Index Sample ID
1 77_1 11 80_3
2 77_2 12 80_4
3 77_3 13 -
4 77_4 14 83_2
5 84_1 15 83_3
6 84_4 16 83_4
7 81_1 17 Zif268
8 81_4 18 FH_bottom
9 80_1 19 CB_top
10 80_2 20 Myc_981
Making more cultures and plates of our expression plasmids
We picked two more colonies from plate 85 today and made 5 ml cultures from them. We also made 2 fresh cultures from the old 85 cultures, by taking 50 ul of old culture and 5 ml LB+spec. We will leave them overnight in the 37* shaker and perform minipreps and PCRs on them tomorrow.
We made positive control cultures from 81.1, 81.4, 80.1, and 80.2, in order to test our minipreps tomorrow. They reactions have a wide range of concentrations post-miniprep, and we are curious to see if the same will happen after performing a miniprep a second time.
Another way to figure out if our expression plasmids contain the appropriate insert is by using a restriction enzyme to cut it and then performing a PCR on the linear DNA. We had previously inserted 2 cutsites on our plasmids, Bsa1 (for insertion of finger 1) and Xba1. The lab already has Xba1, and we ordered Bsa1. Xba1 can be affected by methylation if it is preceded by 'GA' or followed by 'TC' [http://www.neb.com/nebecomm/products/faqproductR0145.asp#874]; we checked and our sequences should not be affected.
We also re-plated 78, 79, and 82 today, one set of plates with 1 ul of culture, and the other set with 10 ul.
Creating the 96 well test plate
Team Wolfe
SNOWMAN!
EcNR2 lambda red (Noah's prep):
- Noah remade the kan-ZFB-wp-hisura insert for us with the kan cassette in the opposite orientation from the his3 and ura3 genes. (The kan cassette is under a strong promoter and does not have a transcriptional terminator, so it's possible that RNA polymerase might keep transcribing through the end of kan and into his3.) He also used it for lambda red in EcNR2, and a couple of plates look like they may have a couple colonies.
- picked 4 colonies, diluted in 20µL H20 and used 1µL as template for 1529620 locus PCR. The rest was grown in LB+kan.
Lambda red with Noah's kanZFBhisura:
- We recently discovered that the kan plates we had been using were very old, and that might be why we got that weird crystalline lawn of colony-like things. We will retry lambda red in the ∆hisB∆pyrF∆rpoZ +pKD46 strain and see if this solves the problem
- 1.5mL cells divided in two: half for kan-his-ura (300ng) and half as a negative control (water)
Thio insert:
- In case lambda red also does not work, we will try to add phosphothioate bases to the insert, but only to the lagging strand 5' end (so only the Ura3 side). Since our primers will have very different melting temps, we will try 2 different reactions:
- Touchdown PCR:
- template: Noah's kan-ZFB-hisura (163ng/µL) diluted to ~10ng per reaction (4 reactions)
- primers: kan_f+homology, Ura3 R phosphothioate
- 2 cycles at each annealing temperature (67, 66...57) for 11 cycles followed by 15 more cycles at 57; 3 min elongation
- Gradient PCR:
- same as above but with 12 reactions, annealing temperatures from 57-67˚C
- Results: most of the gradient reactions produced a faint but clean band at 3kb. The touch down PCR had a stronger 3kb band but also lots of side products.
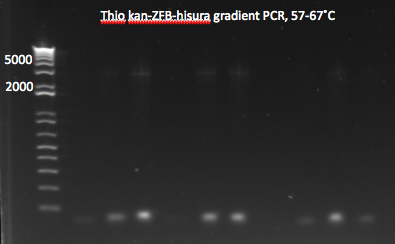
Thio kan-ZFB-hisura (Noah's) gradient part 1 7/18/11
Team TolC
Primers
- Created and ordered primers in order to test the presence of MAGE rpoZ knockout oligo
- Primers made are named
- rpoZ_MAGE_KO plus 4, rpoZ_MAGE_KO minus 4, which have all 4 of the stop codons
- rpoZ_MAGE_KO plus 2, and rpoZ_MAGE_KO minus 2, which have only the first two stop codons.
Kan Plates
- Performed control experiment on Kan plates by growing up liquid culture from glycerol stock and then spreading on Kan plate in order to determine the speed of growth and intensity of the presence of Kan on the plates
- Will check on these tomorrow.
Kan-ZFB-wp insertion
- Picked 5 colonies from ZF090 and ZF091 plates for a total of 10, ZF092 had too few colonies to pick from
- Placed colonies in 20 μL of water and used 1 μl for a 20 μL PCR reaction
- Then took 10 μL from 20 μL dilution and added to 100 μL of LB in 96 well plate and put in 30°C fridge, at 3:19 pm
- Same protocol as KAPA PCR with the following changes made
- Step 3, gradient for annealing, 55°C - 77°C, for a range of 55,57,59,61 and 63°C for the two rows of 5 PCR tubes each (placed leftmost)
- Step 4, Extension, 1 min
- Step 5, 25 cycles total
- Name: TOLCGRAD, PCR5 machine.
July 19th
Team ZF
Checking on plates 78, 79, 82
Both the 10 ul and 1 ul essentially had a lawn of bacteria growing on them. We re-plated these samples, using 1/100 ul and 1/10000 ul of bacteria in 50 ul of LB. We will leave these plates to incubate overnight at 37*.
We also made a culture from the old 85 culture, which we will plate tomorrow.
Minipreps of colonies and controls
Our minipreps from Saturday had low yields, especially for culture 85. We suspect this is why the PCR [[#Gel analysis of miniprep junction PCR on Sunday did not work for this sample. Unfortunately, 85.3 and 85.4 did not grow even after being left in the shaker overnight. These were two cultures we made by picking new colonies from plate 85. This may have occurred because we did not actually pick the colony, or the colony was a cheater. Thus, we performed minipreps on 80.1, 80.2, 81.1, 81.4 (the controls, chosen for their wide range of concentrations after our first miniprep), and 85.1 and 85.2.
In order to figure our why our minipreps were resulting in unexpectedly low yields and increase our yields:
- We tested the pH of our water and Buffer EB.
- The pH of our aliquot of water was ~4-5 (too acidic...we are unsure why)
- The pH of our Buffer EB was ~7.
- Ideally, the DNA should be eluted by a solution of pH 7, so we decided to use Buffer EB during the elution step.
- We also checked the copy number of the origin of replication used in our plasmids. ColE1 apparently has a low copy number, which might also explain our low yields.
- We had made 5 ml of culture in an attempt to increase our yields.
Our yields were larger than our first round of minipreps, although the purities were rather low. We decided to proceed anyway.
 Concentration and purity of our miniprep product |
PCR of miniprep products
We then ran a PCR for the cross-junction on all 6 (80.1, 80.2, 81.1, 81.4, 85.1, 85.2) samples, using our usual cross junction protocol.
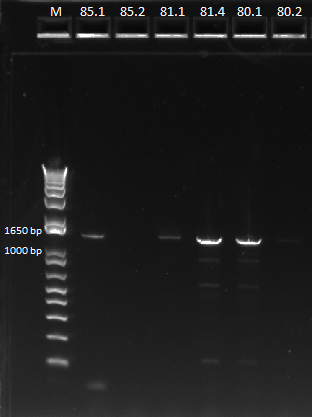 Gel results, expected product size of 1.4 kb |
We saw bands for samples 80.1, 81.1, 81.4, 85.1, and a very faint band for 85.2. The strange thing about these bands is that they appeared to be right around 1650 bp, larger than our 1.4 kb target size. We are unsure of the identity of these bands and will send them out for sequencing.
Starting a new plate for sample 85
Since we have been having such bad luck with sample 85, we seeded 2 ml of LB+spec with 100 ul of the transformed bacteria for sample 85. This is in preparation for making a new plate of sample 85 (the old plate was not a good spec plate and had lots of unspecific growth as well as very few colonies, so if we used this plate for more colony picking our choices would be very limited). This new plate should also give us more luck with picking colonies and having them actually grow in the LB+spec.
Designing ultramers for Zif268, OZ052, and OZ123
In preparation for designing our 96-well practice plate, we designed ultramers for our three positive controls Zif268, OZ052, and OZ123. These ultramers contain the Type II binding sites as well as the F2/F3 fingers. Since the practice plate will contain oligos that encode for F1, we can use these three positive controls to practice and ensure that we secure the technique of using the Type II binding sites to swap in F1 into our expression plasmids. The ultramers have not yet been ordered.
Team TolC
PCR to test insertion of KAN-ZFB-wp into ECNR2
- Made 26 dilutions of culture from three different recombineering plates
- 10 cultures from Kan_tolC_locus_F1 (ZF090 plate) primer recombination
- 10 cultures from Kan_tolC_locus_F2 (ZF091 plate) primer recombination
- 5 cultures from Kan_tolC_locus_F3 (ZF092 plate) primer reecombination
- 1 culture from an old Kan plate with recombination from last week
- 5 of ZF090, 6 of ZF091, and 5 ZF092 were placed in a PCR with 58˚C annealing temperature
- 5 of ZF090, 4 of ZF091, and 1 from old plate were run in a PCR with 62˚C annealing temperature
- primers used were TolC_seq_F and TolC_seq_R
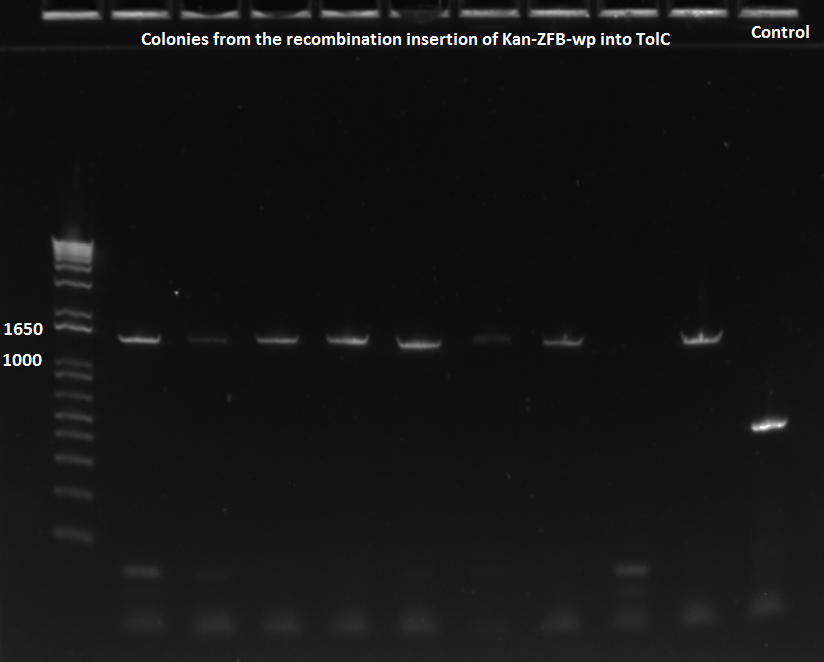
EcNR2 lambda red 1529620 PCR 7/19/11

EcNR2 lambda red 1529620 PCR 7/19/11
- Chose colonies based on the intensity of the bands and grew up cultures of ZF091-2d, ZF091-4d, ZF090-1d, ZF091-5d, ZF090-1s, ZF090-2s, ZF090-3s, and ZF091-3s
Sequencing of Kan-ZFB-wp insertion
- Prepared 16 PCR products of the 8 cultures we chose to grow up in order to send out for sequencing
- Used TolC_Seq_F and ZFB-wp-F primers
- Bands looked a little small when ran out gel for longer
New MAGE oligo
- Constructed a new oligo for MAGE knockout of rpoZ, with oligos more than 4 mutations and substitutions the efficiency drops from 15% down to below 5%, so we created a rpoZ knockout oligo with only 2 substitutions
- The new MAGE rpoZ knockout is rpoZ_MAGE_Subs
- Also ordered primers to test the presence of the knockout rpoZ_MAGE_Subs1+, rpoZ_MAGE_Subs1-, rpoZ_MAGE_Subs2+, rpoZ_MAGE_Subs2-
Team Wolfe
EcNR2 lambda red colony PCR results:
- PCR showed that colonies 1-3 had the insert while 4 did not. Yay! (Note that in the gel image, we accidentally cut off the 300bp band in lane 4)
- We will grow up a reinoculated culture of colony 1 for a glycerol stock. If the Wolfe strain keeps not working, we at least have the insert in an EcNR2 line and can hopefully MAGE out the other genes without too much difficulty.

EcNR2 lambda red 1529620 PCR 7/19/11
pSR01 growth phenotype:
- We will try to used the plate reader again to compare the growth phenotypes of ∆HisB∆pyrF∆rpoZ and ∆HisB∆pyrF∆rpoZ+pSR01. Used the same media as last time but since we're using a 48 well plate, each well has 250µL media. We used 5 pSR01 colonies and one selection strain colony as a control.
- N.B.: this plate we are using is not necessarily meant for OD tests like this and may give some variable readings.
Lambda red results:
- Yesterday's lambda red, which used Noah's kan-ZFB-wp-hisura, has yet to produce colonies. We will leave the plates in longer in case any come up later.
Thio kanZFB:
- Since yesterday's lambda red may not have worked, we need to get the thio kan up and running. We ran out the touchdown PCR samples (since they have more DNA) and will gel extract.
- 57.7ng/µL 260/280=1.98
- 69.3ng/µL 260/280=1.98
July 20th
Team ZF
Checking on 1/100 ul and 1/10000 ul 78, 79, and 82 plates
We got colonies on all the plates, but the 1/100 ul plates look a bit crowded. On the other hand, the 1/10000 plates only have a few colonies. We decided to save both plates, and decide which to use later.
- Based on these results, a 1/1000 ul dilution might be best.
Check on 85 culture
There was no growth in our culture.
Later: It ends up that our ultramer from 85 (Pos Con 77) had an error in it. Instead of coding for fingers 2 and 3, it codes for fingers 1 and 2. We ordered a new ultramer, and we no longer need to work on 85.
Finalizing the practice plate
We decided on using a practice plate with 24 oligos, consisting of 18 target sequences, and 6 controls. Our 6 controls are Zif268, OZ052, OZ123, and 3 sequences from the CODA table. We will need to construct the expression plasmid for Zif268, OZ052, and OZ123. Their ultramers were ordered today.
Our 18 targets were GNN/TNN sequences, which are more likely to have a binder, and were distributed among the backbones. The target and control groups each have their own set of primers, to facilitate picking them from the pool.
Expression Plasmid Sequencing Results
We got back our sequencing results:
| Name | ID | Final? | Notes
|
| FH Top (77) | 1 | N | 1 snp
|
| | 2 | N | 5 snps
|
| | 3 | Y | G -> C. Still Ala
|
| | 4 | N | 1 snp
|
| FH Bottom (78) | 1 | N | 1 Deletion at 1746, last nucleotide of F of FQRICMRN
|
| CB Top (79) | 1 | N | A lot of deletions/mistakes in f2/f3 region
|
| CB Bottom (80) | 1 | Y |
|
| | 2 | Y | G->C. Stiil Ala
|
| | 3 | N | deletion 1613-1631
|
| | 4 | N | many mistakes at end, may be ok
|
| Myc 198 (81) | 1 | Y |
|
| | 4 | Y | G->C. Still Ala.
|
| Myc 981 (82) | 1 | Y |
|
| Pos Con 16 (83) | 4 | Y | G->C. Still Ala.
|
| | 2 | N | A lot of deletions/mistakes in f2/f3 region
|
| | 3 | N | A lot of deletions/mistakes in f2/f3 region
|
| Pos Con 55 (84) | 1 | Y (?) | Right sequence, but messy trace
|
| 4 | N | at 547: deletion at type II binding site
at 665: deletion at beginning of F3
|
| Zif 268 | 1 | Y |
|
| Summary:
|
| Good: 77.3, 80.1, 80.2, 81.1, 81.4, 82, 83, 84.1 (?), Zif268
|
| Bad (Redo): 78, 79
|
We picked 4 new colonies for 78, and 79 since their sequences had many errors. We left these cultures overnight at 37* in the shaker.
Construction of OZ052 and OZ123: Joining the Finger with Homology regions
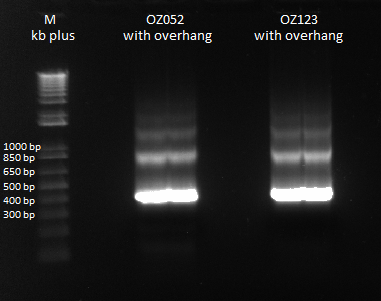
Previously, we did a PCR to reassemble the ultramers for OZ052 and OZ123, but they only coded strictly for F1/F2/F3 and did not have any overhang homology with any surrounding code. Thus, we performed PCR to add these homology overhangs so that we can integrate them with the omega subunit as well as the spec backbone to create a final expression plasmid.
The program we used was 55* annealing temperature, and a 15 second extension time. Gel results pictured below.
 Overlap PCR to extend ultramers with homologies |
We had an expected product size of 400 bp., which we can see in the gel. Bands are a little weird since there are bands heavier than the heaviest theoretical band (which is the strong product). We will do a gel extraction tomorrow to get rid of these bands to obtain pure product for use in the isothermal assembly.
Team TolC
rpoZ MAGE knockout
- Ran two rounds of MAGE on 4 ECNR2 cultures with the kan-ZFB-wp insertion using Noah's rpoZ knockout oligo
- Grew cultures up to mid-log in kan+amp and LB
- Four cultures are ZF091-2d, ZF091-4d, ZF090-1d, and ZF090-5d
- Electroporator read 5.4 ms for all four cultures in first round
- Could not do a third round of MAGE since the E.Coli do not appear to have recovered, and are nowhere near midlog (MAGE round 2 was done by 5:15pm and the cultures were checked at 8:30pm). Will let the culture go on overnight.
- Put 50ul of a 1:100 dilution of each culture on Amp plates. Used a spreader for plates 1&2 and glass beads for plates 3 & 4.
Glycerol Stocks
- Made 4 glycerol stocks of the cultures sent to be sequenced
- ZF090-1s, ZF090-2s, ZF090-3s, ZF091-3s
convinced that the gradient PCR machine is out of order, ought to inform Andrew
Team Wolfe
IPTG Plate and the Plate Reader
- Technical delays have allowed us only a few hours worth of plate reading for a 46-well plate with selection strain + pSR01 in LB+Amp +/- IPTG, NM+His +/- IPTG, NM +/- IPTG, and NM+3-AT +/- IPTG. Strains used: ∆HisB∆pyrF∆rpoz+pSR01 (5 colonies) and ∆HisB∆pyrF∆rpoz (1 colony).
- The plate had a noticeable white residue under the lid around around the edges. We believe this was due to flaking of the plastic cover from vibration in the plate reader.
- Cell growth in the plate suggested that IPTG is killing our cells since few cultures survived with IPTG and those that did (LB+Amp) grew less than the equivalent cultures without IPTG.
New Plate Without IPTG
- On plate reader at 11:30AM
- Strains used: Water as control, Selection (Wolfe) Strain, Selection Strain + pSR01
- Media Used: LB+Tet (LB+Amp for SS+pSR01),NM+His, NM, NM+3-AT (1µM), NM+3-AT (2.5µM), NM+3-AT (5µM).
Lambda Red
Same procedure as usual
- Added ~350ng of Thio-Kan-ZFB-wp-His3-Ura3
- Used "new cuvettes"
- Negative water control
Plate Reader Update
TEMPORARILY OUT OF SERVICE
Your worst nightmare
http://www.youtube.com/watch?v=v4o8MDjuu-A
July 21st
Team ZF
Checking colonies 78 and 79
All of the cultures grew overnight, so we performed a miniprep on them to obtain the plasmids.
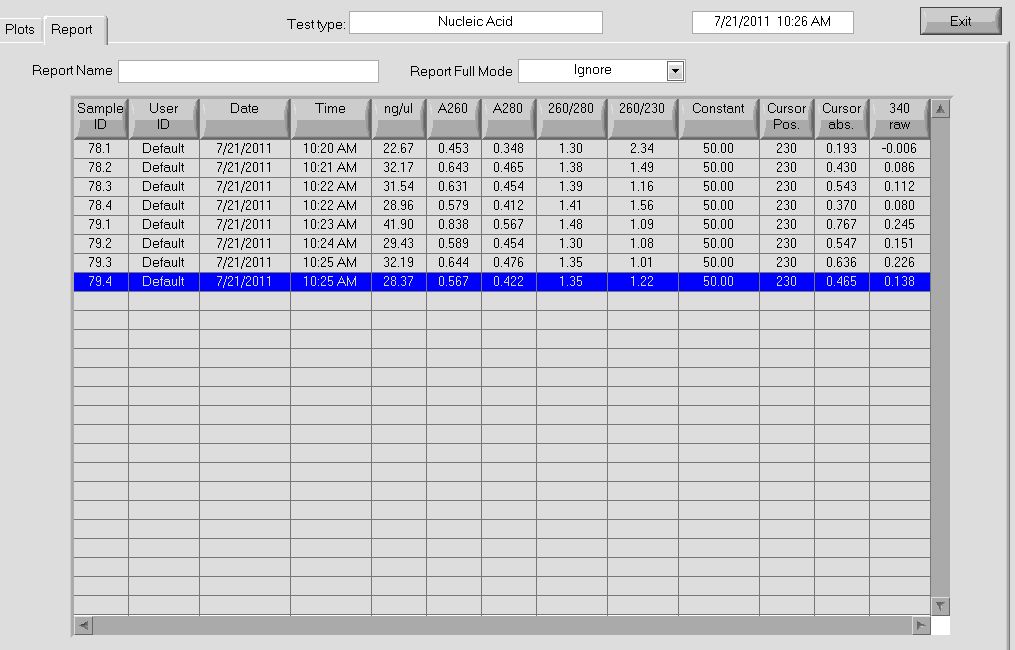 Plasmid concentrations after miniprep |
Our concentrations were better than they have been in the past; notably, after the addition of buffer P2, our solution actually turned a bright blue (indicating lysis of the cells), which we had not seen before.
We then ran a PCR on our miniprep product, to check for our desired insert. We used our usual protocol for a PCR of the expression plasmid cross-junction.
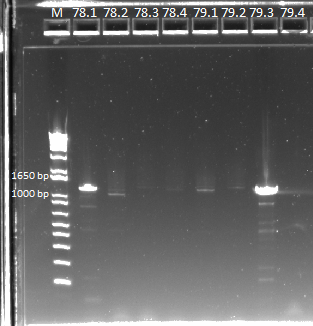 PCR results, note that some lanes do not show strong bands |
Some of the products did not show up very strongly, but we sent them out for sequencing in case it was a PCR/gel error.
Sending out our expression colonies for sequencing
We sent out the samples from yesterday that had no more than 1 SNP for reverse sequencing. In addition, we also sent out 78 and 79 for forward and reverse sequencing.
- Reverse sequencing: 77.1, 77.3, 77.4, 78.1, 80.1, 80.2, 80.4, 81.1, 81.4, 82.1, 83.4, 84.1, 84.4, Zif268
- Forward and reverse sequencing: 781, 78.2, 78.3, 78.4, 79.1, 79.2, 79.3, 79.4
For sequencing, we used the forward primer pZE23G_3581_F CCAACCTTACCAGAGGGCGC (Junction forward) and the reverse primer pZE23G_2123_R CGAACGACCTACACCGAACT (Junction reverse)
We did a PCR of the sequences from yesterday that we want sequenced again.
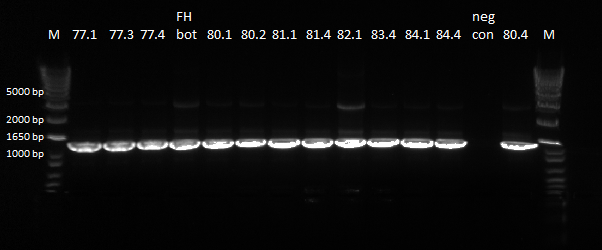 PCR results, looking good |
The bands were clear, and we will send these products to be sequenced.
Gel Extraction and Isothermal Assembly of OZ fingers+homology
We ran a gel extraction of our PCR product from yesterday, so that we could use it for isothermal assembly later. Strangely, we got bands that were larger than our largest theoretical product.
 Prepare yourself to be gel-extracted |
We had the following yields from our gel extraction:
ID Conc. (ng/ul) 260/280
OZ052 96.3 1.93
OZ123 125.5 1.91
Based on these concentrations, we used the following recipes for our isothermal reaction:
- OZ052:
- Spec: 0.63 ul
- OZ052: 1.56 ul
- Omega: 0.13 ul
- Water: 1.56 ul
- Isothermal assembly solution: 7.5 ul
- OZ123:
- Spec: 0.63 ul
- OZ123: 0.14 ul
- Omega: 0.13 ul
- Water: 1.60 ul
- Isothermal assembly solution: 7.5 ul
We left the isothermal assembly product in the -20* freezer, and will perform the transformation tomorrow.
Team Tolc
Team Wolfe
- Lambda red culture did not grow colonies overnight on a kanamycin plate. This may be due to a low time (4.8ms) during the electroporation.
- Repeated lambda red using the same procedure. Got a similar time constant and noticed that the water negative control had more cells than the culture containing the insert, but we went ahead and plated 1mL of each on kan plates.
- Also replated the 7/20 lambda red culture to see if we can get any good cells (1.5mL).
July 22nd
Team ZF
Sequencing Results of Expression Plasmids
The good news is that we managed to get a perfect sequence for each of our plasmids. The bad news is that we do not know which sequences are the matches, since the names are all scrambled for some reason (i.e. sample 78.x does not align with FH bottom like it should, but instead aligns perfectly with another plasmid sequence). We will thus need to resend our samples for sequencing to be able to choose the correct plasmids that match. Below is a summary of the scrambled data from the sequencing we sent in yesterday:
| ID
| Is a perfect match with
| Notes
|
| Myc 198 (81) | 78.2, 78.3 |
|
| FH Top (77) | 81.4, Myc 981 | 83.4 has 1 deletion
|
| CB Bottom (80) | 84.4 | 78.1 has 5 deletions; Zif has 1 deletion
|
| FH bottom (78) | n/a | 84.1 deletion at beginning of F3
|
| CB Top (79) | 79.3 |
|
| Myc 981 (82) | 78.4 |
|
| Zif 268 | 79.4 |
|
| Pos Con 55 (83) | 79.2 | 79.3 has deletion at beginning of F3
|
| Pos Con 16 | 79.1 |
|
To this end, we redid the PCRs for everything in preparation for the resequencing, using the junction primers and the previous protocol. The resulting gels can be observed below:
 E-gel showing bands for samples 78.1-78.4 and 79.1-79.4, expected size of 1.4 kb |
 Remaining samples, expected size of 1.4 kb |
From these PCRs, all the reactions look like they produced bands of the right size, a good indicator that the isothermal assemblies worked. Only sequencing will tell whether they are truly the products that we are looking for.
We sent these samples out for sequencing, making sure that all the samples were in the appropriate tubes.
Transformation for OZ plasmids
Today, we took the results of the isothermal assembly from yesterday and did chemical transformation to introduce the plasmids into the Top 10 ChemComp bacteria, using our protocol for chemical transformations. They were left to recover for approximately 1.5 hours. We plated 2 dilutions, 10 ul and 50 ul, and left them overnight at 37* in the incubator.
Team Wolfe
- Yesterday's lambda red plate had several large colonies--but so did the negative control plate. We will go ahead and do a PCR of the 1529620 locus (as previously described) on 12 of the colonies in case one of them happens to have the insert. The colonies were also grown up simultaneously in LB+kan.
- Results: all of the colonies were cheaters
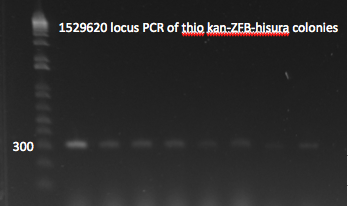
1529620 locus PCR 7/22/11

1529620 locus PCR part 2 7/22/11
- In case these colonies do not have the insert, as we suspect, we will repeat lambda red with the thio kan-ZFB-wp-his3-ura3 insert
- 1mL culture for each condition, 210 ng DNA
- time constant for thio insert sample was 5.0
Team TolC
Gel for Jun's MAGE rpoZ, rowD
- The gel appeared to have bands, so we are running a second PCR to confirm it
 "
"
.png)

_and_omega-ultramer_plasmid_pcr_annotated.png)





.png)
.png)




.png)







.png)
.png)