From 2011.igem.org
(Difference between revisions)
|
|
| (3 intermediate revisions not shown) |
| Line 147: |
Line 147: |
| | ===Team ZF=== | | ===Team ZF=== |
| | ====PCR to check for omega+zif268==== | | ====PCR to check for omega+zif268==== |
| - | All colonies that grew on the plates we left overnight should have spec-resistance, but they may not have the omega and zif268 unit that we inserted [[#Isothermal Assembly|yesterday]] with isothermal assembly. We performed a PCR on 13 colonies picked from the 50 ul plate to find a colony that contains the w+zif268 insertion. We used the same recipe at our PCR reaction [[#PCR of omega+zif268 with overhangs|on Friday]]. If the insert is present, then we should see a band at 709 bp. We also included 2 positive controls: our isothermal assembly product and our selection strain from which the w+zif268 was originally copied. | + | All colonies that grew on the plates we left overnight should have spec-resistance, but they may not have the omega and zif268 unit that we inserted yesterday with isothermal assembly. We performed a PCR on 13 colonies picked from the 50 ul plate to find a colony that contains the w+zif268 insertion. We used the same recipe at our PCR reaction [[#PCR of omega+zif268 with overhangs on Friday. If the insert is present, then we should see a band at 709 bp. We also included 2 positive controls: our isothermal assembly product and our selection strain from which the w+zif268 was originally copied. |
| | | | |
| | We ran an agarose gel to check the bands: | | We ran an agarose gel to check the bands: |
| Line 155: |
Line 155: |
| | |} | | |} |
| | | | |
| - | None of the colonies displayed any bands, except for primer residues. Lane 16 contained the selection plasmid, on which we performed [[#Gel purification of PCR product|a successful PCR]] on Friday, so we definitely expected a band at 709 bp. Because the positive controls did not have a band at 709 bp, we suspect that something went wrong with the PCR. We plan on re-running the PCR tomorrow.</div> | + | None of the colonies displayed any bands, except for primer residues. Lane 16 contained the selection plasmid, on which we performed [https://2011.igem.org/Team:Harvard/Protocols#PCR_purification a successful PCR] on Friday, so we definitely expected a band at 709 bp. Because the positive controls did not have a band at 709 bp, we suspect that something went wrong with the PCR. We plan on re-running the PCR tomorrow.</div> |
| | <div id="711" style="display:none"> | | <div id="711" style="display:none"> |
| | + | |
| | ==July 11th== | | ==July 11th== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 272: |
Line 273: |
| | | | |
| | ===Team Web=== | | ===Team Web=== |
| - | *Design [https://spreadsheets.google.com/spreadsheet/ccc?key=0ApTl36bX3P7qdFo3SEFXMkNCZ3ZBX1hBQUZqWUpiV2c&pli=1#gid=0 Google Doc]</div> | + | *Looked at good wikis from last year. </div> |
| | <div id="712" style="display:none"> | | <div id="712" style="display:none"> |
| | + | |
| | ==July 12th== | | ==July 12th== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 294: |
Line 296: |
| | |} | | |} |
| | | | |
| - | Even after the gel purification, our bands were streaky. We decided to redo the PCR, placing all the reactions in single tubes, performing a touchdown PCR instead. Our annealing temperature started at 70*, repeated for 2 cycles, and then decreased 2*, every two cycles, until it reached 58* (the optimal annealing temperature), where it remained until the 25 cycles were complete. We left this PCR going overnight. (See [[#Results of ultramer touchdown PCR|July 13th]] for the gel image.) | + | Even after the gel purification, our bands were streaky. We decided to redo the PCR, placing all the reactions in single tubes, performing a touchdown PCR instead. Our annealing temperature started at 70*, repeated for 2 cycles, and then decreased 2*, every two cycles, until it reached 58* (the optimal annealing temperature), where it remained until the 25 cycles were complete. We left this PCR going overnight. (See July 13th for the gel image.) |
| | | | |
| | ====PCR of expression plasmid cross-junction==== | | ====PCR of expression plasmid cross-junction==== |
| | | | |
| - | We also performed a second PCR on colony nine (the colony we decided with which we decided to proceed) to confirm that our omega+zif268 in the spec backbone was successfully inserted into the ''E. coli''. The expected product was the cross-junction on the plasmid, approximately 1.4 kb. We used the same recipe as that used on [[#PCR of omega+zif268 with overhangs|July 9th]], except with a forward primer of PZE23G-3581 F and a reverse primer of PZE23G-2133 R, and had a 55* annealing temperature and an extension time of 45 seconds. | + | We also performed a second PCR on colony nine (the colony we decided with which we decided to proceed) to confirm that our omega+zif268 in the spec backbone was successfully inserted into the ''E. coli''. The expected product was the cross-junction on the plasmid, approximately 1.4 kb. We used the same recipe as that used on July 9th, except with a forward primer of PZE23G-3581 F and a reverse primer of PZE23G-2133 R, and had a 55* annealing temperature and an extension time of 45 seconds. |
| | | | |
| | After running an e-gel, we confirmed that our w+zif268 was inserted into the plasmid. | | After running an e-gel, we confirmed that our w+zif268 was inserted into the plasmid. |
Latest revision as of 18:48, 3 August 2011
July 7th
Team ZF Assembly
Our primers still haven't arrived. We are working with the Web Design Team.
Team Wolfe Selection
- PCR for Round 2 MAGE cultures in LB media
- The samples came from the overnight culture of MAGE/control strains grown in LB+amp, NM+his, and NM. Oddly, the (∆HisB)∆pyrF∆rpoZ+pKD46 control grew only in NM+His and LB, and not in plain NM.
- Samples
- Control= 1 ss+pKD46
- 11 cultures from 1μL plate in LB media
- 12 cultures from 10μL plate in LB media
- Program
- Anneal at 56°
- Elongation for 90 seconds
- All else according to standard KAPA protocol
- E gel of control and 1µL plate colonies 1-8 to check PCR worked (it was successful)
- samples prepared for sequencing tomorrow
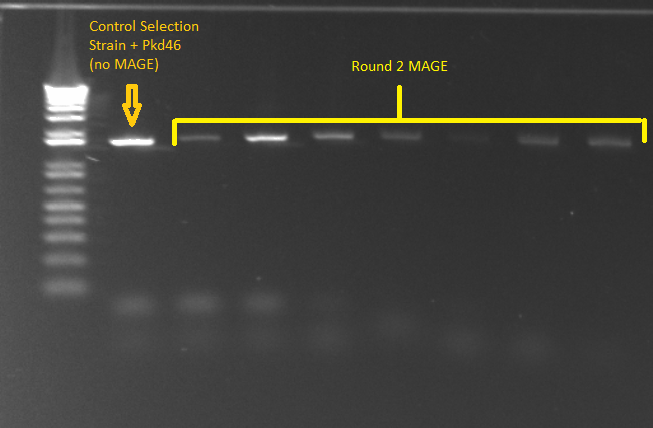
MAGE 2 samples for sequencing 7/7/11
- Kan-ZFB insert check PCR results:
- the overnight cultures grown yesterday were used in a PCR (same KAPA protocol as before) around the 1529620 locus. Not all the cultures grew; 100µL plate cultures 1-5 and 2mL plate cultures 1-12 were used.
- Results: some of the reactions, strangely, did not work, but the ones that did showed a short band and thus did not have the insert (see below)
- HisBNuke3 MAGE round 3:
- used same protocol as previously described, using 1.5mL mid-log culture from MAGE round 2
- will recover overnight so that we can reinoculate and continue MAGE rounds tomorrow
- NM medium:
- added 3.9g glucose to 100mL NM medium to bring the glucose concentration up to the correct level.
- Because (∆HisB)∆pyrF∆rpoZ+pKD46 did not grow in plain NM, we are curious to see if that was due to low glucose levels. One colony from a plate was put in 3mL NM(gluc) and will be grown overnight.
- HisB MAGE2:
- In case the sequencing does not show successful MAGE, we will grow overnight 48 more colonies (24 from each plate) in 100µL of NM(gluc) and LB+amp
Team TolC
- making culture tubes of ECNR2 and ECNR2delTolC for glycerol stock (no Amp)
- making culture tubes of ECNR2 for MAGE to delete rpoZ (one with amp, one without)
- dilutions of rpoZ deletion MAGE oligo, 20µM
- dilution of rpoZ deletion MAGE oligo, 1µM, for MAGE.
- serial dilution of ECNR2 after MAGE round 1, and round 2, with 20µl of 100x dilution of concentrated culture. For plating onto amp plates.
- During Gel extraction, ignored 3x volume in step 2 and instead used 500 µl of QG buffer. Additionally, used 18 µl of EB buffer and not water in step 13
Web Design
Brainstorming
- Minimalist toolbar, metallic crimson color
- "Popout" menu below, not dropdown
- Slideshow on front page
- First slide: Brief text project description, brief youtube video description (in layman's terms)
July 8th
Team ZF Assembly
PCR of omega+zif268 with overhangs
We performed a PCR to take out our omega subunit plus zif268. We used the following recipe:
- 10 ul KAPA readymix
- 0.75 ul of hindIII (primer)
- 0.75 ul of wF+plLacO (primer)
- 1.2 ul template
- 12.3 ul water
The first steps of the PCR involved heating at 95* (for 5 minutes) and 98* (for 20 seconds). Annealing temperature was 56*. Extension time was 30 seconds.
The product of the PCR should be 709 bps. It includes the omega subunit, zif268, as well as overlap sequences homologous to the selection plasmid.
We ran an e-gel to ensure that our desired product was present:
Gel purification of PCR product
We had to gel purify our PCR product in preparation for isothermal assembly, using our protocol for gel purification. The result from running the gel can be seen below - we cut out the bands at 709 bp, the expected size of our product.

In the final step of the purification process, we eluted our DNA in buffer EB. We used the nanodrop spectophotometer to determine the concentration of DNA present.
- The concentration was 54.3 ng/ul.
Team Wolfe Selection
MAGE round 3:
- MAGE cultures that recovered overnight were saturated, so we made 1:1000, 1:10000, and 1:100000 dilutions and plated 20µL of each onto amp plates and left at 30˚C overnight.
MAGE round 2 cultures from yesterday:
- We did a PCR (His_F and and His_R primers) on 24 of the 48 cultures we grew and sent them off for sequencing.
Results of NM culture:
- The selection strain (∆HisB∆pyrF∆rpoZ+pKD46) did not grow in NM, which is worrisome. None of the MAGE samples grew in NM either, so either they all have HisB knocked out or there is something wrong already with the strain's His pathway.
- We will grow up selection strain (without pKD46) from glycerol stock in LB+tet and transform with Vatsan's plasmid tomorrow to see if that has any effect on the his-deficiency phenotype.
Kan-ZFB insertion: modified nucleotides:
- Primers finally arrived that would put modified nucleotides on the 5' ends of the insert, which should improve lambda red efficiency.
- PCR: KAPA protocol with Kan forward phosphorthioate and URA2 reverse phosphorthioate, 56˚C annealing, 90 sec elongation, 25 cycles, 1µL kan-ZFB-hisura DNA as template
- Results: PCR only produced a smaller side product. This is strange since we also ran a gel of all our different kan-ZFB-hisura purifications and they all run at the correct size (see images below).

kan-ZFB insert purifications
Team TolC
- Performed re-inoculation of ECNR2-Placed into tube labeled ECNR 2 MAGE 3 in 30 degree shaker.
- grew up 24 colonies total for sequencing ECNR2 for rpoZ deletion (the colonies appear to have grown much faster than expected).
- rows A (MAGE round 1) and B (MAGE round 2), for PCR and sequencing. to be placed in the 4C refrigerator after use as template.
- RPOZ PCR-Machine 6
- Primer Annealing = 56 degrees
- Extension 48 = seconds
Team Web Design
- Edited/Re-drafted our project description, see Dropbox (It's a bit long as is now, but the excess informaiton that we cut out can still probably be used somewhere on our website
- Sketched draft pages of the public wiki
- Homepage- first draft sketch done based on the characteristics and requirements we'd discussed. However, there are a few issues (i.e., how to include some of the iGEM wiki reuqirements).
July 9
Team ZF
Isothermal Assembly
We performed an isothermal assembly in order to insert omega+zif268 into the plasmid containing the spec backbone.
- The concentration of the plasmid (previously prepared) was 157.4 ng/ul.
- The concentration of the w+zif268 was 54.3 ng/ul.
Using our isothermal assembly protocol we determined we needed:
- 0.63 ul backbone (length: ~2.2 kb)
- 0.59 ul w+zif268 (length: ~0.7 kb)
- 2.78 ul water
- 15.0 ul isothermal assembly solution
Transformation of electro-competent E. coli
We electroporated our cells so they would take up the plasmids, using:
- 1.0 ul DNA
- 40.0 ul cells
- 500 ul LB
We plated the cells, using this protocol, on spec plates, one of which contained 50 ul of cells and the other 150 ul of cells. The cells have been incubated at 37*, and will be left there overnight. Cells that have taken up the plasmid have spec-resistance and should be able to survive. Tomorrow, we will be picking colonies and performing PCR on them to determine whether they contain w+zif268.
Team Wolfe
- MAGE3 plates look good--we'll choose colonies for sequencing on Monday.
- Transformation of selection strain with Vatsan's plasmid:
- We're curious to see whether Vatsan's plasmid (Zif268 omega, ZFB, wp, hisura) will have an effect on the (∆HisB)∆pyrF∆rpoZ strain's growth despite the seemingly intact HisB.
- Transformed mid-log selection strain (reinoculated from overnight culture) with about 100ng of Vatsan's plasmid following standard procedure.
- After 2 hr recovery plated on amp plates with 100µL or 1mL
July 10th
Team ZF
PCR to check for omega+zif268
All colonies that grew on the plates we left overnight should have spec-resistance, but they may not have the omega and zif268 unit that we inserted yesterday with isothermal assembly. We performed a PCR on 13 colonies picked from the 50 ul plate to find a colony that contains the w+zif268 insertion. We used the same recipe at our PCR reaction [[#PCR of omega+zif268 with overhangs on Friday. If the insert is present, then we should see a band at 709 bp. We also included 2 positive controls: our isothermal assembly product and our selection strain from which the w+zif268 was originally copied.
We ran an agarose gel to check the bands:
None of the colonies displayed any bands, except for primer residues. Lane 16 contained the selection plasmid, on which we performed
a successful PCR on Friday, so we definitely expected a band at 709 bp. Because the positive controls did not have a band at 709 bp, we suspect that something went wrong with the PCR. We plan on re-running the PCR tomorrow.
July 11th
Team ZF
We did a miniprep of the 12 colonies we picked Sunday and left overnight in LB. Here are the concentrations of our products:
| ID
| Conc. (ng/ul)
| 260/280
|
| 1
| 6.63
| 1.27
|
| 2
| 12.96
| 1.53
|
| 3
| 9.28
| 1.41
|
| 4
| 7.23
| 1.31
|
| 5
| 12.64
| 1.55
|
| 6
| 10.77
| 1.48
|
| 7
| 17.19
| 1.73
|
| 8
| 39.70
| 1.78
|
| 9
| 55.73
| 1.81
|
| 10
| 24.34
| 1.72
|
| 11
| 25.19
| 1.68
|
| 12
| 38.57
| 1.74
|
| 13
| 27.26
| 1.75
|
Then we performed a PCR on the 12 miniprep products, the 12 colonies in LB, as well as 3 controls (negative control, isothermal assembly product, selection strain) to find a colony that has the omega+zif26 insertion:
- 10 ul KAPA readymix
- 0.75 ul of hindIII (primer)
- 0.75 ul of wF+plLacO (primer)
- 1.2 ul template
- 12.3 ul water
The first steps of the PCR involved heating at 95* (for 5 minutes) and 98* (for 20 seconds). Annealing temperature was 56*. Extension time was 30 seconds.
We ran a 1% 100 ml. agarose gel at 120V to check the success of the PCR.
 Gel of our colonies, miniprep-ed colonies, and controls to check for w+zif268 insertion |
The bands were rather thick, so it was difficult to estimate the exact band size. However, the bands appeared a bit below 650 bp, although we expected a size of 709 bp, so we ran an e-gel on some of the colonies to ensure that that bands were indeed the right size.
 Gel to ensure our bands were the correct size (709 bp) |
The bands were closer to the size that we expected, so we decided that we successfully inserted w+zif268 into the plasmid. We decided to pick colony 9 (which had the highest concentration post-miniprep) and work with that from now on. We made spec plates with 50 ul of colony 9.
Team Wolfe
Selection strain transformed with pSR01:
- both plates had lots of colonies--almost too many. We need to make sure the plasmid is, indeed, in these cells.
- Colony PCR:
- picked 6 colonies and diluted in 7µL H2O: 3 for PCR template and 4 to be grown up in LB+amp
- KAPA protocol using M13_F and M13_R primers (anneal to the vector), 50˚C annealing, 2min elongation
- E gel results: lots of bands but rather messy and not exactly what we'd expect. We'll try to verify the presence of the plasmid in a few other ways.
- Sequencing:
- We sent out the PCR samples with M13_R to Genewiz
- Culture PCR:
- We used the cultures made with the colonies used for PCR above as template to see if this gives us cleaner results.
- same protocol as above except with 1µL of 1:20 dilution of culture as template.
- Assuming that the plasmid is indeed in the colonies, we will grow an overnight culture in LB+amp as well as the Wolfe selection strain and tomorrow we will use a growth reader to check their phenotypes in NM
MAGE round 3 colonies:
- In case pSR01 does not rescue the selection strain's growth phenotype, we will PCR 96 colonies from the MAGE3 plates and send them out for sequencing tomorrow.
- Used 9µL KAPA, His_F and His_R primers, 3µL suspended colony (out of 7--the remaining 4 grown in LB+amp)
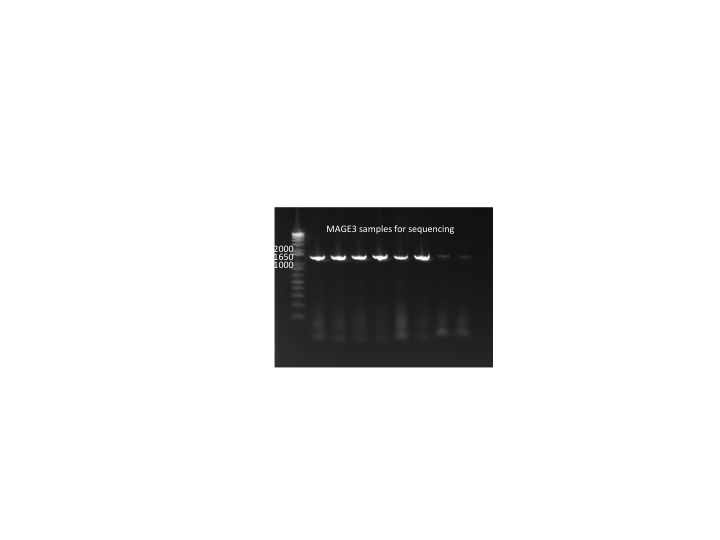
MAGE3 colonies for sequencing 7/11/11
Thio kanZFB:
- We retried the PCR to put modified nucleotides on the ends of the kan-ZFB-wp-hisura insert using the same procedure as 7/8/11
- E gel: reactions didn't work
- Tried again with a longer elongation time (3 min) and let run overnight.
Team TolC
Sequencing of rpoZ delete
- Since the sequencing was sent after 3pm on Friday, it wasn't picked up before today (Monday), so we are going ahead with lambda red recombineering of the KAN-ZFB-wp into the ECNR2 genome
- glycerol stocks of ECNR2 and ECNR2ΔTolC were made.
Team Web
- Looked at good wikis from last year.
July 12th
Team ZF
PCRs of finger ultramers
We received the ultramers for fingers two and three of our 6 experimental expression plasmids. We performed an overlap PCR in order to join the two ultramer pieces and make a whole unit. Ultimately this unit with be assembled with the omega subunit and the spec backbone to make our final expression plasmids. We used the following recipe:
- 10 ul KAPA
- 13.5 ul water
- 0.75 ul forward ultramer
- 0.75 ul reverse ultramer
We had an annealing temperature of 58* and an extension time of 15 seconds.
 Gel of our product from the ultramer PCR. Note that the first lane's PCR was done in a single tube, while all the others were done in a strip. |
However, in addition to the expected band at approximately 300 bp, the bands for our ultramers was extremely streaky, indicating that there may be other contaminants. We decided to perform a PCR purification of these samples, using our protocols, and see if there was an improvement in the gel.
 Gel of our product after PCR purification |
Even after the gel purification, our bands were streaky. We decided to redo the PCR, placing all the reactions in single tubes, performing a touchdown PCR instead. Our annealing temperature started at 70*, repeated for 2 cycles, and then decreased 2*, every two cycles, until it reached 58* (the optimal annealing temperature), where it remained until the 25 cycles were complete. We left this PCR going overnight. (See July 13th for the gel image.)
PCR of expression plasmid cross-junction
We also performed a second PCR on colony nine (the colony we decided with which we decided to proceed) to confirm that our omega+zif268 in the spec backbone was successfully inserted into the E. coli. The expected product was the cross-junction on the plasmid, approximately 1.4 kb. We used the same recipe as that used on July 9th, except with a forward primer of PZE23G-3581 F and a reverse primer of PZE23G-2133 R, and had a 55* annealing temperature and an extension time of 45 seconds.
After running an e-gel, we confirmed that our w+zif268 was inserted into the plasmid.
 Lane 3: Gel of our PCR to check for the cross-junction in colony 9. |
PCR and gel extraction of omega subunit
We performed a PCR to get out the omega subunit+linker, with an annealing temperature of 56* and an extension time of 15 seconds. The miniprep from colony 8 was the template DNA. We will use the product in assembling our final expression plasmids.
We determined that the PCR was successful after running the sample on an agarose gel, and performed a gel extraction so that we could use the product in our isothermal assembly.
Lanes 1 and 2: Gel of our PCR to get omega subunit. |
The gel extraction was successful, and we had a final concentration of 120.5 ng/ul.
Overlap PCR of OZ052 and OZ123
We performed an overlap PCR using the ultramer pairs that solely encoded the F1, F2, and F3 fingers of OZ052 and OZ123. The annealing temperature was 62* and the extension time was 15 seconds. The results of the PCR can be found below (the products are only in the last two lanes) and the products were also PCR purified before being put into storage at -20*C.
 Last two lanes: OZ052 and OZ123 products, respectively |
Team Wolfe
MAGE round 3 samples:
- 94 sent out for sequencing (numbers go A1-H1, etc.)
Thio kan-ZFB results:
- PCR run out on E Gel: not successful. We do not know what is still causing our PCR to fail.
pSR01-transformed selection strain:
- We discovered that hisB is a two-function protein, with the N-terminal region acting as an HPase and the C-terminal region as an IDPG. His3 only complements the IDPG activity, so even if our MAGE worked, it might destroy our selection system. The two aa deleted in hisB are in the C terminal region, so maybe they do inactivate it. We emailed Keith Joung whose lab made the selection strain to see how HisB was deleted.
- In case this mutation is correct but lambda red in the selection strain still eludes us, we designed MAGE oligos for use in EcNR2 to either delete the two aa or put 3 stop codons after the HPase region.
- culture PCR results: cultures 3-4 had a strong band (and 2 and 5 weak ones) around 850-1000bp. It is interesting how different these results are from yesterday's colony PCR gel.

pSR01 culture and thio kan-ZFB PCRs 7/12/11
- Sequencing results: The sequences sent from Genewiz aligned well with the pSR01 plasmid sequence and covered pretty much all of URA3. This implies that the cells did indeed take up our plasmid.
- Growth phenotype:
- 12 colonies were chosen and split between NM (gluc), NM+IPTG (to induce zif268 expression), NM+his (gluc), NM+his+IPTG, LB+amp, LB+amp+IPTG and grown overnight. (Final concentration IPTG=500µM)
Team TolC
- MAGE knockout sequencing
- Received sequencing from the MAGE knockout of rpoZ within the TolC genome
- Observed all 24 sequenced attempts and it appears as none of them have the knockout and stop codons placed into genome
- Possible reason behind failure is that one, we may not have been reading the results correctly or two, the MAGE oligo had all three stop codons right next to each other in the oligo and may have needed to be separated by three nucleotides each in order to maximal annealing
- Lambda Red Recombination of kan-ZFB-wp insertion into MAGE knockouts
- Plates from the recombination showed small colonies
- With this we picked 6 colonies from the 4 different plated recombination cultures and grew them to saturation
- Began PCR with TolC_Seq_F and TolC_Seq_R primers in order to determine whether recombination was successful
- Procedure (on gradient PCR machine, name: ABC, folder: igem)
- KAPA mixture
- Then 95 C, 5 min
- 98˚ C, 20 sec
- 60˚ C, 15 sec
- 75˚ C, 45 sec
- Repeated cycle for 30 times
- 72˚ C, 5 min
- 4˚ C, forever
- Tomorrow we plan to run gel
- Length will be:
- 450 bp if insertion failed
- 1200 bp if insertion of Kan cassette only
- 1650 bp if insertion of Kan-ZFB-wp
- Insertion Construct for TolC (homology-kan-ZFB-wp-homolgy)
- Began 90 µL total of PCR for the insertion construct since we ran out
- If gels show failed PCR tomorrow we will start from normal ENR2 without any MAGE and begin with lambda red recombination and possibly make new MAGE primer and then knockout rpoZ
Team Web Design
- Met with Harvard biosafety officer, Sidney
- Send to him: list of strains, plasmids that we are using
- Note to all iGEM-ers: We will all be attending a biosafety seminar on Friday, July 15th from 10am to 12n in Biolabs 1058.
- In case you haven't already, please see Alain's message below:
- Every member of the Harvard iGEM team (unless you are absent on Friday) has to attend the next lab Safey/Biosafety Blood born pathogens training this coming Friday 7/15/2011 from 10 am to 12 noon. The training takes place in the Biological laboratories (16 Divinity Ave) room 1058. If you cannot go and have a very good reason let me (Alain) know. If you attended a similar training session on the Cambridge campus or at the Medical school you don't have to attend. Otherwise you have to go.
July 13th
Team ZF
Results of ultramer touchdown PCR
We ran an e-gel on the product of our touchdown PCR. Once again we have our band at 344 bp. It is still slighly streaky, but much less than our earlier PCR reaction. There are some faint bands smaller than our expected product, which are likely the ultramers we used in the reaction. We'll use the product for our isothermal assembly later today.
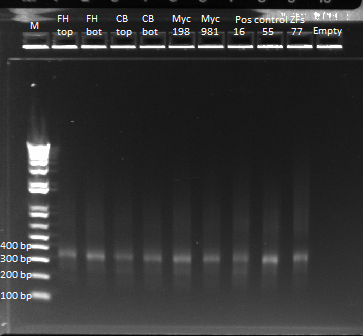 Gel of our ultramer touchdown PCR product |
Isothermal assembly and transformation of expression plasmids
We followed our protocol for isothermal assembly, using the following recipe:
- 0.63 ul backbone (at 157.4 ng/ul to get 100 ng)
- 0.13 ul omega+linker (at 120.5 ng/ul)
- 0.50 ul ultramer (~30 ng/ul)
- 1.24 ul water
- 7.5 ul isothermal assembly solution
The sizes of the pieces we put together were the following:
- Backbone: 2.2 kb
- omega+linker: 352 bp
- ultramer: 344 bp
Note: We also made a negative control of the isothermal assembly, only putting in the spec backbone without omega+linker or ultramer.
We then performed transformations to insert our isothermal assembly products into E. coli, following our usual protocol. However, we ran out of electrocompetent cells, so only samples 78 - 82 were done with electroporation while 77, 83-85, and the negative control were done with chemical transformation, using these protocols (the samples are named after the reverse ultramer ID in the primer index that were used to generate the ultramer sequences).
We then plated 1 ul of the cells on spec plates and left them overnight at 37*. Because we ran out of our own spec plates, we also had to use LB plates that had spec added to it, and spec plates that were made in April.
Team Wolfe
∆HisB∆pyrF∆rpoZ+pSR01 growth results:
- When no IPTG was present, the bacteria grew in LB+amp, NM+his, and NM, showing that the plasmid rescues the selection stain's growth phenotype! This implies that our strain had the right genotype all along. However, when IPTG was added to the media, the strain only grew well in LB. This is very odd, since IPTG was supposed to induce zif268 and thus His3 expression. We will try a couple things to make sure nothing went wrong:
- redo cultures in the same pattern as yesterday--perhaps IPTG+/- were mislabeled
- inoculate cultures of ∆HisB∆pyrF∆rpoZ +/- pSR01 so that Vatsan can use a plate reader to compare their growths
- Joung wrote back and said that the HisB deletion is in fact 6 bases in frame. He also mentioned something about a lac deletion which we are looking into.
MAGE3 sequencing results:
- none of the colonies had the stop codons. This may be because mutS is not knocked out, and it can correct strands that have up to 5 bases mismatched.
Kan-ZFB for lambda red:
- Noah did lambda red in EcNR2 and the plate was covered in crystalline patterns that may be cells. We will test this by:
- Restreaking some of these "colonies" onto a fresh kan plate to get the bacteria more spread out
- Streaking a kan plate with pKD46 (from glycerol stock) to see if the kan is effective
- Taken a streak off the plate, diluted it in 200µL H20, and used 1µL as template for PCR
- KAPA, 1529620-flanking primers, 90 sec elongation and 56 anneal, 25 cycles
- Results: 300bp band--the streaks on the plate are bacteria, but they do not have the insert.

1529620 locus PCR of EcNR2 lambda red 7/13/11
- We also retried the thio kan-ZFB PCR using some new primer aliquots. Once again it failed.
- Because this PCR keeps failing, we're going to try a different way and do two separate and shorter PCRs to be joined later in an overlap PCR:
- PCR of kan cassette: kan cassette as template (diluted 6/16 aliquot), kan F phosphothio and kan R primers, KAPA, 56˚C anneal and 90 sec elongation
- PCR of ZFB-wp-hisura: ZFB-wp-hisura as template (diluted 6/16 aliquot), ZFB-wp-f and URA3 R phosphothio primers, KAPA, 60˚C anneal and 90 sec elongation
Team TolC
- PCR ECNR2
- Ran PCR on four colonies from lambda red recombination results, ECNR2 liquid culture from Church lab, and colony from original ECNR2 plate
- Grew each up in LB Amp liquid culture
- Tomorrow the gel will tell us whether the insertion was successful and also if the ECNR2 cultures are pure
- PCR Noah's MAGE on rpoZ knockout
- Picked 15 colonies from each plate with ECNR2 without TolC and ECNR2 with TolC
- Created liquid cultures of all 30 colonies picked and growing up over night
- PCR will be ready to send out tomorrow for sequencing
- More kan-ZFB-wp insertion construct needed
- Ran PCR in order to obtain more kan-ZFB-wp insertion construct from the kan-ZFB-wp-His3-PyrF construct
- Placed on gel and used gel extraction in order to purify

KAN-ZFB-wp construct with tolC homology 7/13/11
Team Web Design
- Safety, safety, safety!
- Received safety confirmation letter
- Finalized project description
- Updated our iGEM wiki page for the upcoming deadlines (Project description and safety, due July 15th)
July 14th
Team ZF
Results of the expression plasmid transformation
Only samples 78 to 82 had colonies after overnight incubation--these were the same colonies that were electroporated, instead of chemically transformed. Because we only added 1 ul of our cells to the plates, it is possible that this was not enough sample for us to see colonies, especially since chemical transformation is not as efficient as electroporation. The samples on the LB+spec plates had what appeared to be unspecific growth around the edges. This may be because the spec did not diffuse evenly. We will be re-plating all of our samples later today, on new LB+spec plates.
PCR of product from isothermal assembly of expression plasmid
We also ran a PCR on the product of the three-part isothermal assembly (omega+linker, ultramers, spec backbone). We used the cross-junction primers and the same recipe as our PCR on Tuesday. Once again we expected a product of 1.4 kb.
 1.2% E-gel of our ultramer isothermal assembly reaction product |
There were fairly sharp bands underneath the band we expected. Additionally, the expected 1.4 kb band was not visible for samples 80 and 81, although they were successfully grown on plates. Sample 85 had no bands, not even primers.
Since 80 and 81 grew on our plates yesterday, we continued with our plan of re-plating our original transformations, especially since the chemically transformed cells do have the appropriate isothermal assembly product to insert. We re-plated all the transformations on the LB+spec plates made today, and will leave them overnight in the 37* incubator. We also made cultures from the colonies on plates 78-82 to mini-prep tomorrow.
Rerunning of PCR products (ultramers and isothermal assembly)
In addition, we decided to see if the streaking was possibly caused by the gel instead of a PCR problem, and so we ran three of our products in a 1% agarose gel instead of an E-gel, with the results below.
 Products ran in an agarose gel. Note that wells 78-80 on the left side of the gel are empty due to sample mysteriously floating out of the well. |
Team Web Design
- Intro video: brief (~1:30) Flash video explaining our project (Our project in 30 sec? 1 min?)
- Interview Joung/Persikov-- videotape (documentary style) for Human Practices page
- Human Practices
- Broader societal impacts
- Gene therapy, personalized medicine
Team Wolfe
thio kan and thio ZFB-wp-hisura PCR:
- Ran overnight PCR of kan cassette and ZFB-wp-hisura on E gel: looks great!

thio kan and thio ZFB-wp-hisura PCR 7/14/11
- PCR purification with Qiagen kit:
- added an additional 300µL buffer PB to adjust the pH
- thio kan cassette: 59.9ng/µL 260/280=1.90
- thio ZFB-wp-hisura: 100.9ng/µL 260/280=1.89
- Overlap PCR:
- 4 rxns: two used undiluted template (60ng thio kan cassette and 101ng thio ZFB-wp-hisura), two used template diluted 1:10
- KAPA, 56˚C anneal, 90 sec elongation, 25 cycles
- primers (kan f phosphothio and ura3 r phosphothio) added after 10 cycles
- Results: PCR worked but with tons of side products so we will try to optimize the procedure. Expected product = 3Kb, notable side products = 2Kb, 4Kb. N.B. The samples were accidentally loaded while the gel was running so they are staggered on the gel.
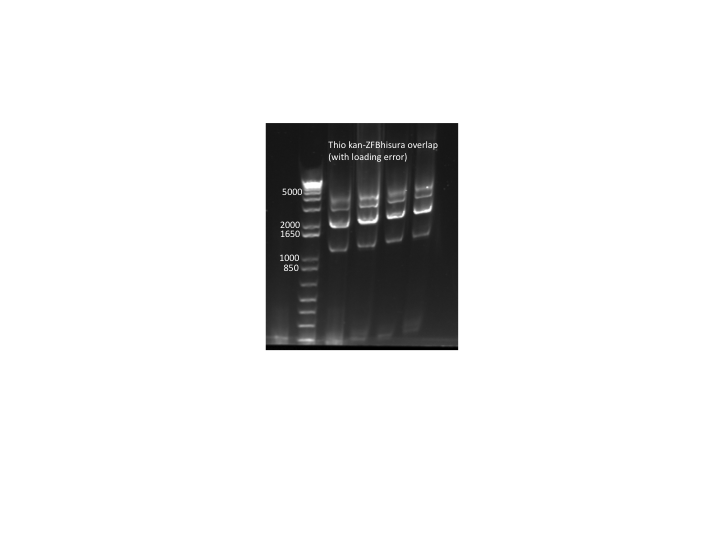
thio kan-ZFB-wp-hisura overlap PCR (with loading error)
- Gel stab PCR
- DNA from the 3Kb e-gel band were removed using the stabbing technique and placed in 4 respective reaction tubes (diluted in 20µL water)
- The 4 samples were separated into template dilutions of 20X, 100X, 200X, and 400X and phosphothio primers for PCR to amplify the desired 3Kb kan-ZFB-wp-hisura sequence. PCR results will be reported tomorrow.
- Template PCR followed by PCR with primers:
- PCR just the template (1µL diluted thio kan cassette and 1µL diluted thio ZFB-wp-hisura) with no primers, KAPA, 12 cycles, 60˚C anneal, 90 sec elongation
- take PCR product and use it as template for new PCR either undiluted, diluted 1:4, 1:16, 1:64, 1:256; kan f phosphothio and ura3 r phosphothio primers, 25 cycles, 59˚C anneal, 90 sec elongation
pSR01 phenotype:
- Just to be sure, we will assemble cultures in a flat-bottomed 96 well plate and use a plate reader to track the growth curves.
- 2 colonies ∆HisB∆pyrF∆rpoZ, 10 colonies transformed with pSR01
- media: LB+amp, NM+his, NM, NM+3-AT (1mM), LB+amp+IPTG(100µM), NM+his+IPTG, NM+IPTG, NM+3-AT+IPTG
- plate reader: absorbency of 600nm, measurements every 10 min, shaking 560 sec before each measurement (settings saved in iGEM folder on computer in tissue culture room)
Team TolC
- PCR done of 4 colonies picked from KAN plate (with ECNR2 after recombineering with the KAN-ZFB-wp construct. Running an E-gel showed that the insert was not present. Since the KAN plates were old, it might be speculated that the plates were old, and so, the colonies were not selected for KAN resistance.
- Primers were TolC_seq_F and TolC_seq_R.
- Without insertion these two primers will create 449 bp sequence, and with insertion length will be 1650 bp

TolC insert check 1 7/14/11
- Some of the original culture after recombineering, 1ml solution spun down, and resuspended in 100μl of LB, and plated on new KAN plates. To check, one of the old KAN plates was plated the same way and put in at 2:30pm. (These plates will be checked tomorrow morning.)
- PCR done of a few of the rpoZ MAGE with stop codon insertions. Oligo name: rpoZ_MAGE_KO (Noah's oligo) (Noah's plates, after two cycles of MAGE, on both ECNR2 and ECNR2ΔTolC). The side product in the gel could be due to a low annealing temperature of 53°C, so we should do a gradient PCR (e.g.55-66°C) to get the most product the next time.
 "
"






.png)
.png)

.png)



_annotated.png)
.png)

.png)
.png)
.png)


.png)
.png)
.png)