"
"
Team:Baltimore/Modeling
From 2011.igem.org
(Difference between revisions)
| (2 intermediate revisions not shown) | |||
| Line 4: | Line 4: | ||
:Baltimore's iGEM Team 2011 is continuing the work begun in 2010. Last year we had a strong DIY focus.We still have that focus, as well as an academic slant to the Team. | :Baltimore's iGEM Team 2011 is continuing the work begun in 2010. Last year we had a strong DIY focus.We still have that focus, as well as an academic slant to the Team. | ||
| - | '''The Taq project''': Our goal has been and is to provide a way for anyone to be able to make their own Taq polymerase instead of having to purchase it. Below is a diagram of the construct made using a program called PlasmaDNA. This program is available as a free download and is a good program for diagramming cloning strategies. The plasmid was never sequenced, but checks out with respect to restriction mapping and produces a protein of the correct size 92 kd that has thermostable polymerase activity. | + | '''Wet-ware:The Taq project''': Our goal has been and is to provide a way for anyone to be able to make their own Taq polymerase instead of having to purchase it. Below is a diagram of the construct made using a program called PlasmaDNA. This program is available as a free download and is a good program for diagramming cloning strategies. The plasmid was never sequenced, but checks out with respect to restriction mapping and produces a protein of the correct size 92 kd that has thermostable polymerase activity. |
:Notice that in our model you can see the restriction enzymes enzyme sites that were used to splice in the Pol1 coding sequence for Taq polymerase. | :Notice that in our model you can see the restriction enzymes enzyme sites that were used to splice in the Pol1 coding sequence for Taq polymerase. | ||
| Line 11: | Line 11: | ||
:The modeling for However the gene for Taq polymerase has a PstI site located within the coding sequence which makes it incompatible with the BioBrick assembly standard. So the first order of business was to change the sequence through site directed mutagenesis in a way that would maintain the amino acid sequence of the protein but change the DNA sequence of the gene. | :The modeling for However the gene for Taq polymerase has a PstI site located within the coding sequence which makes it incompatible with the BioBrick assembly standard. So the first order of business was to change the sequence through site directed mutagenesis in a way that would maintain the amino acid sequence of the protein but change the DNA sequence of the gene. | ||
| - | |||
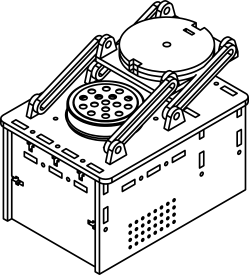
| - | + | '''Dry-ware:The Equipment''': | |
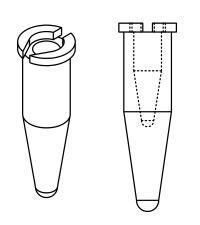
| - | + | :PCR Tube Containers: We have designed spin holders for PCR tubes, which make it possible to use the regular centrifuge, without a purchased insert for spinning small PCR tubes. The model was drawn using SolidWorks.<br>[[File:PCRShim.png]] <br> | |
| - | + | :PCR Machine: We have also been working on an inexpensive PCR machine. The model was drawn using SolidWorks.<br>[[File:Bmore_CheapPCR_Design.png]] <br> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
Latest revision as of 03:48, 29 September 2011
| Home | Team | Project | Parts Submitted to the Registry | Modeling | Notebook | Solutions | Safety | Attributions | Hardware | Material Safety Data Sheets | [http://partsregistry.org/assembly/libraries.cgi?id=35|MIT Parts Registry] |
|---|
- Baltimore's iGEM Team 2011 is continuing the work begun in 2010. Last year we had a strong DIY focus.We still have that focus, as well as an academic slant to the Team.
Wet-ware:The Taq project: Our goal has been and is to provide a way for anyone to be able to make their own Taq polymerase instead of having to purchase it. Below is a diagram of the construct made using a program called PlasmaDNA. This program is available as a free download and is a good program for diagramming cloning strategies. The plasmid was never sequenced, but checks out with respect to restriction mapping and produces a protein of the correct size 92 kd that has thermostable polymerase activity.
- Notice that in our model you can see the restriction enzymes enzyme sites that were used to splice in the Pol1 coding sequence for Taq polymerase.

- The modeling for However the gene for Taq polymerase has a PstI site located within the coding sequence which makes it incompatible with the BioBrick assembly standard. So the first order of business was to change the sequence through site directed mutagenesis in a way that would maintain the amino acid sequence of the protein but change the DNA sequence of the gene.
Dry-ware:The Equipment:
- PCR Tube Containers: We have designed spin holders for PCR tubes, which make it possible to use the regular centrifuge, without a purchased insert for spinning small PCR tubes. The model was drawn using SolidWorks.

- PCR Machine: We have also been working on an inexpensive PCR machine. The model was drawn using SolidWorks.