|
|
| (8 intermediate revisions not shown) |
| Line 2: |
Line 2: |
| | ==June 24== | | ==June 24== |
| | *Designed primer for testing HisB deletion, reuse His_Internal_R to test the band | | *Designed primer for testing HisB deletion, reuse His_Internal_R to test the band |
| | + | |
| | + | '''pZE21G:''' |
| | + | *reinoculated culture with 100µL of saturated solution, grew to mid-log, and made glycerol stock |
| | + | *backbone PCR: ran E gel but no bands--PCR unsuccessful. We may need to use a different backbone for the zinc fingers. |
| | + | |
| | + | '''Omega and Omega+Zif268:''' |
| | + | *these were the only two PCR reactions from 6/22/11 to work |
| | + | *PCR purified using Qiagen kit: |
| | + | **omega: 6.1ng/µL, 260/280=1.83 |
| | + | **omega+Zif268: 11.3 ng/µL, 260/280=1.67 |
| | + | |
| | + | '''Lambda red recombination of selection system:''' |
| | + | *reinoculated selection strain+pKD46 with 100µL of saturated solution |
| | + | *just before mid-log (about 4 hours after inoculation) divided culture in half (1.5mL) and added either 37.5µL or 3.75µL of 20% arabinose solution (to try two different induction levels). Cultures grew for another hour. |
| | + | *The rest of the procedure was the same as the 6/22/11 attempt but without the 42C water bath. |
| | + | |
| | + | ==June 24th - Bioinformatics== |
| | + | ===Playing with Pseudocounts=== |
| | + | |
| | + | Using CTC because of position 6's reliance on the CNN frequencies, we see what difference values of pseudocounts (if in the frequency table, the frequency of an amino acid is 0, bump it up to the psuedocount: ex. A = 0 becomes A = .015 with a psuedocount of .015) make. Pseudocounts are necessary for data that has small sample size - we could be missing out on working helices because a letter's frequency is 0 when it shouldn't be. |
| | + | |
| | + | Various pseudocount (psu = ) values. Look at the 7th column, which is position 6 in the helix: |
| | + | |
| | + | {| |
| | + | | [[File:HARVCTC_0.png|thumb|left|psu = 0]] |
| | + | | [[File:HARVCTC_.005_psuedo.png|thumb|left|psu = .005]] |
| | + | | [[File:HARVCTC_.008_psuedo.png|thumb|left|psu = .008]] |
| | + | |- |
| | + | | [[File:HARVCTC_.01.png|thumb|left|psu = .01]] |
| | + | | [[File:HARVCTC_.015_psuedo.png|thumb|left|psu = .015.]] |
| | + | | [[File:HARVCTC_.02_psuedo.png|thumb|left|psu = .020.]] |
| | + | |} |
| | + | |
| | + | The variation from E being the top letter to A being top back to E is from a slight adjustment in how we add on psuedocounts: the 'new' way is a more proportional approach. |
| | + | |
| | + | Notice how psu = 0 gives only the four letters found in our dataset, while psu > 0 adds in other letters, each with a small probability ranging from .5% to 2%. |
| | + | |
| | + | The question is how much psu to add: less means we weight our (possibly flawed) data of proven zinc fingers more. Higher psu adds more randomness (variation) to our sequences, but some fraction of those sequences will not work. |
| | | | |
| | ===Updated Closest Zif268 Fingers=== | | ===Updated Closest Zif268 Fingers=== |
| | We realized that some of our "close non-zif268 fingers" were actually not all that close to Zif268, and so we went into the 88,000 zinc finger database and pulled out zinc fingers surrounding zif268. In fact, there were many, many, many zinc fingers that had identical sequences to the Zif268 F2 finger, and so we looked at sequences around it. The tree below shows the new non-zif268 backbones that are actually close to zif268 compared to our old set. The new set is in gray, the old set is in black. This gives us a potential seven more backbones to work with. | | We realized that some of our "close non-zif268 fingers" were actually not all that close to Zif268, and so we went into the 88,000 zinc finger database and pulled out zinc fingers surrounding zif268. In fact, there were many, many, many zinc fingers that had identical sequences to the Zif268 F2 finger, and so we looked at sequences around it. The tree below shows the new non-zif268 backbones that are actually close to zif268 compared to our old set. The new set is in gray, the old set is in black. This gives us a potential seven more backbones to work with. |
| | [[File:HARVComparisonTree.png]] | | [[File:HARVComparisonTree.png]] |
| - | ==June 24th - Bioinformatics==
| |
| - |
| |
| | ===Sequence Generation=== | | ===Sequence Generation=== |
| | We made some small updates to the sequence generator, based on the frequencies we noticed in the outputs of the tests we ran. | | We made some small updates to the sequence generator, based on the frequencies we noticed in the outputs of the tests we ran. |
| Line 50: |
Line 86: |
| | **Conservative distribution 56.3 : 32.8 : 10.9 | | **Conservative distribution 56.3 : 32.8 : 10.9 |
| | **Riskier distribution 33.3 : 33.3 : 33.3 | | **Riskier distribution 33.3 : 33.3 : 33.3 |
| - |
| |
| - | ==June 24th==
| |
| - | '''pZE21G:'''
| |
| - | *reinoculated culture with 100µL of saturated solution, grew to mid-log, and made glycerol stock
| |
| - | *backbone PCR: ran E gel but no bands--PCR unsuccessful. We may need to use a different backbone for the zinc fingers.
| |
| - |
| |
| - | '''Omega and Omega+Zif268:'''
| |
| - | *these were the only two PCR reactions from 6/22/11 to work
| |
| - | *PCR purified using Qiagen kit:
| |
| - | **omega: 6.1ng/µL, 260/280=1.83
| |
| - | **omega+Zif268: 11.3 ng/µL, 260/280=1.67
| |
| - |
| |
| - | '''Lambda red recombination of selection system:'''
| |
| - | *reinoculated selection strain+pKD46 with 100µL of saturated solution
| |
| - | *just before mid-log (about 4 hours after inoculation) divided culture in half (1.5mL) and added either 37.5µL or 3.75µL of 20% arabinose solution (to try two different induction levels). Cultures grew for another hour.
| |
| - | *The rest of the procedure was the same as the 6/22/11 attempt but without the 42C water bath.
| |
| - |
| |
| - | ==June 24th - Bioinformatics==
| |
| - | ===Playing with Pseudocounts===
| |
| - |
| |
| - | Using CTC because of position 6's reliance on the CNN frequencies, we see what difference values of pseudocounts (if in the frequency table, the frequency of an amino acid is 0, bump it up to the psuedocount: ex. A = 0 becomes A = .015 with a psuedocount of .015) make. Pseudocounts are necessary for data that has small sample size - we could be missing out on working helices because a letter's frequency is 0 when it shouldn't be.
| |
| - |
| |
| - | Various pseudocount (psu = ) values. Look at the 7th column, which is position 6 in the helix:
| |
| - |
| |
| - | {|
| |
| - | | [[File:HARVCTC_0.png|thumb|left|psu = 0]]
| |
| - | | [[File:HARVCTC_.005_psuedo.png|thumb|left|psu = .005]]
| |
| - | | [[File:HARVCTC_.008_psuedo.png|thumb|left|psu = .008]]
| |
| - | |-
| |
| - | | [[File:HARVCTC_.01.png|thumb|left|psu = .01]]
| |
| - | | [[File:HARVCTC_.015_psuedo.png|thumb|left|psu = .015.]]
| |
| - | | [[File:HARVCTC_.02_psuedo.png|thumb|left|psu = .020.]]
| |
| - | |}
| |
| - |
| |
| - | The variation from E being the top letter to A being top back to E is from a slight adjustment in how we add on psuedocounts: the 'new' way is a more proportional approach.
| |
| - |
| |
| - | Notice how psu = 0 gives only the four letters found in our dataset, while psu > 0 adds in other letters, each with a small probability ranging from .5% to 2%.
| |
| - |
| |
| - | The question is how much psu to add: less means we weight our (possibly flawed) data of proven zinc fingers more. Higher psu adds more randomness (variation) to our sequences, but some fraction of those sequences will not work.
| |
| | | | |
| | '''List of Remaining Goals:''' | | '''List of Remaining Goals:''' |
| Line 97: |
Line 94: |
| | *Yay</div> | | *Yay</div> |
| | <div id="625" style="display:none"> | | <div id="625" style="display:none"> |
| | + | |
| | ==June 25th-26th - Bioinformatics== | | ==June 25th-26th - Bioinformatics== |
| | | | |
| Line 156: |
Line 154: |
| | | | |
| | ===100 Control Sequences=== | | ===100 Control Sequences=== |
| - | * See our [[File:HARVPositive Control Sequences PostMacro.xlsx]], updated June 28th | + | * See our [https://static.igem.org/mediawiki/2011/5/5d/HARVPositive_Control_Sequences_PostMacro.pdf Positive Control Sequences], updated June 28th |
| | * Selected known binding zinc fingers from the CODA table that bind sequences similar to our target sequences | | * Selected known binding zinc fingers from the CODA table that bind sequences similar to our target sequences |
| | * All control helices from CODA were inserted into Zif268 F2 backbones and have been assigned a seventh primer tag separate from the tags given to the 6 target sequences. | | * All control helices from CODA were inserted into Zif268 F2 backbones and have been assigned a seventh primer tag separate from the tags given to the 6 target sequences. |
| Line 184: |
Line 182: |
| | | | |
| | ===Cut Site Design=== | | ===Cut Site Design=== |
| - | *See our [[Cut Site Design]] page | + | *See our [https://2011.igem.org/Team:Harvard/Cut_Site_Design Cut Site Design] page |
| | *We left in one proline (P) between the linker and the starting FCQ... of finger 2, but as this proline is the last AA of the OPEN linker (TGEKP) and occurs before the beta sheet in every zinc finger in Zif268 (see zif268's sequence on its [http://www.pdb.org/pdb/explore/remediatedSequence.do?structureId=1AAY PDB page]) | | *We left in one proline (P) between the linker and the starting FCQ... of finger 2, but as this proline is the last AA of the OPEN linker (TGEKP) and occurs before the beta sheet in every zinc finger in Zif268 (see zif268's sequence on its [http://www.pdb.org/pdb/explore/remediatedSequence.do?structureId=1AAY PDB page]) |
| | *This configuration also allows the library to be used at any finger position because proline ends the OPEN linker. | | *This configuration also allows the library to be used at any finger position because proline ends the OPEN linker. |
| Line 232: |
Line 230: |
| | <font color=red>'''''Attention all Harvard iGEM-ers!!!'''''</font> <font color=blue> According to the [https://2011.igem.org/Main_Page iGEM Main Page], our preliminary project descriptions and safety proposals are due on</font> <font color=red>'''''July 15'''''</font>. <font color=blue> Please see the aforementioned link so we can get this done ASAP- we don't want to miss any deadlines and have all our hard work wasted!</font> | | <font color=red>'''''Attention all Harvard iGEM-ers!!!'''''</font> <font color=blue> According to the [https://2011.igem.org/Main_Page iGEM Main Page], our preliminary project descriptions and safety proposals are due on</font> <font color=red>'''''July 15'''''</font>. <font color=blue> Please see the aforementioned link so we can get this done ASAP- we don't want to miss any deadlines and have all our hard work wasted!</font> |
| | | | |
| - | | + | *Finalized our [https://static.igem.org/mediawiki/2011/5/5d/HARVPositive_Control_Sequences_PostMacro.pdf Positive Control Sequences], using Justin's macro to insert the F1 helices into the appropriate zif268 F2 backbone |
| - | *Finalized our [[File:HARVPositive Control Sequences PostMacro.xlsx|Positive Control Sequence Table]], using Justin's macro to insert the F1 helices into the appropriate zif268 F2 backbone | + | |
| - | | + | |
| | | | |
| | *Length of chip oligos: 131-140bp (based on [[Cut Site Design]]) | | *Length of chip oligos: 131-140bp (based on [[Cut Site Design]]) |
| Line 242: |
Line 238: |
| | **Type II binding/cut sites= 11bp on each side (22bp total) | | **Type II binding/cut sites= 11bp on each side (22bp total) |
| | **Standard legnth: 40 + 69 + 22 = 131bp | | **Standard legnth: 40 + 69 + 22 = 131bp |
| - |
| |
| | | | |
| | *Use WebLogos as a final visual check of our final generated sequences | | *Use WebLogos as a final visual check of our final generated sequences |
| - |
| |
| | | | |
| | ===Plasmid and Oligo Design Schematics=== | | ===Plasmid and Oligo Design Schematics=== |
| Line 263: |
Line 257: |
| | | | |
| | ===Harvard Logo=== | | ===Harvard Logo=== |
| - |
| |
| | {| | | {| |
| | | [[File:HARVHarvard_logo.png|thumb|left|]] | | | [[File:HARVHarvard_logo.png|thumb|left|]] |
| | |} | | |} |
| - |
| |
| - |
| |
| | ===Running the Generator!=== | | ===Running the Generator!=== |
| | [[File:HARVFasta_total.csv]] NOTE: LATER GENERATED NEW SEQUENCES. NOT UP TO DATE. | | [[File:HARVFasta_total.csv]] NOTE: LATER GENERATED NEW SEQUENCES. NOT UP TO DATE. |
| Line 276: |
Line 267: |
| | **During this time, we created a function that will re-translate the sequences that the generator output. It compares the original helix with the re-translated helix to make sure that our reverse-translate works properly. | | **During this time, we created a function that will re-translate the sequences that the generator output. It compares the original helix with the re-translated helix to make sure that our reverse-translate works properly. |
| | ***This step went smoothly, and we verified that the sequences were reverse-translated properly. | | ***This step went smoothly, and we verified that the sequences were reverse-translated properly. |
| - | **To make sure that the distributions generated were as expected, we made [[#Generated WebLogos for Final Chip|WebLogos]] of the helices generated(see below). | + | **To make sure that the distributions generated were as expected, we made WebLogos of the helices generated(see below). |
| | *The output file (in the Dropbox: iGem > chip > final chip.csv) originally had the following headers: 'Target', 'Backbone #', 'Helix Sequence', 'Backbone Sequence', 'Nucleotide Sequence of Zinc Finger' | | *The output file (in the Dropbox: iGem > chip > final chip.csv) originally had the following headers: 'Target', 'Backbone #', 'Helix Sequence', 'Backbone Sequence', 'Nucleotide Sequence of Zinc Finger' |
| | **We wanted to convert this information into FASTA format. | | **We wanted to convert this information into FASTA format. |
| Line 295: |
Line 286: |
| | | [[File:HARVTGG.png|thumb|left|TGG]] | | | [[File:HARVTGG.png|thumb|left|TGG]] |
| | |} | | |} |
| - |
| |
| | | | |
| | *FASTA-Formatted Chip Data: | | *FASTA-Formatted Chip Data: |
| Line 308: |
Line 298: |
| | |} | | |} |
| | | | |
| - | | + | [[File:HARVPrimer_Index_iGEM_2011.xls]] |
| - | [[File:HARVPrimer Index_iGEM 2011]] | + | |
| | | | |
| | ===Design of Plate Practice Sequences=== | | ===Design of Plate Practice Sequences=== |
| Line 328: |
Line 317: |
| | | | |
| | The primer tag sequences for the 90 generated sequence subset will be the same as they are on the chip (for the sake of explanation, we will refer to them now as P1F and P1R in this paragraph). The positive controls will be flanked immediately by the same primers as the generated subset so that we can amplify everything as one pool altogether should we need to (so this will be P1F and P1R). However, we will also put an additional set of primers outside of the P1F/P1R primers for the positive controls so that we can specifically amplify the positive control subpool, should we want to. These primers will be the same as the primers for the positive control on the chip (which will be called P2F and P2R here). | | The primer tag sequences for the 90 generated sequence subset will be the same as they are on the chip (for the sake of explanation, we will refer to them now as P1F and P1R in this paragraph). The positive controls will be flanked immediately by the same primers as the generated subset so that we can amplify everything as one pool altogether should we need to (so this will be P1F and P1R). However, we will also put an additional set of primers outside of the P1F/P1R primers for the positive controls so that we can specifically amplify the positive control subpool, should we want to. These primers will be the same as the primers for the positive control on the chip (which will be called P2F and P2R here). |
| - |
| |
| | | | |
| | To recap, on the chip we will have the following oligos : | | To recap, on the chip we will have the following oligos : |
| Line 343: |
Line 331: |
| | Oligo (+ control, 6 total): | P2F | P1F | type II binding site | generated F1 | type II binding site | P1R | P2R | | | Oligo (+ control, 6 total): | P2F | P1F | type II binding site | generated F1 | type II binding site | P1R | P2R | |
| | </pre> | | </pre> |
| - |
| |
| | | | |
| | Once we get our test sequences back from IDT, they will come in a 96-well plate with one oligo in each plate. We should make a mixture using some of each well in order to create a tube that contains all 96 sequences. This will simulate the tube that we will receive from Agilent, except instead of 55,000 sequences we will have 96 sequences only in this tube. From here, we can practice using this as a library. | | Once we get our test sequences back from IDT, they will come in a 96-well plate with one oligo in each plate. We should make a mixture using some of each well in order to create a tube that contains all 96 sequences. This will simulate the tube that we will receive from Agilent, except instead of 55,000 sequences we will have 96 sequences only in this tube. From here, we can practice using this as a library. |
| Line 353: |
Line 340: |
| | We will be repeating these exact same steps once we get the chip, so if we can perfect our protocols with these practice sequences, we should be golden when the chip comes in.</div> | | We will be repeating these exact same steps once we get the chip, so if we can perfect our protocols with these practice sequences, we should be golden when the chip comes in.</div> |
| | <div id="629" style="display:none"> | | <div id="629" style="display:none"> |
| | + | |
| | ==June 29th== | | ==June 29th== |
| | Our first day with everyone in the wet lab! | | Our first day with everyone in the wet lab! |
June 24
- Designed primer for testing HisB deletion, reuse His_Internal_R to test the band
pZE21G:
- reinoculated culture with 100µL of saturated solution, grew to mid-log, and made glycerol stock
- backbone PCR: ran E gel but no bands--PCR unsuccessful. We may need to use a different backbone for the zinc fingers.
Omega and Omega+Zif268:
- these were the only two PCR reactions from 6/22/11 to work
- PCR purified using Qiagen kit:
- omega: 6.1ng/µL, 260/280=1.83
- omega+Zif268: 11.3 ng/µL, 260/280=1.67
Lambda red recombination of selection system:
- reinoculated selection strain+pKD46 with 100µL of saturated solution
- just before mid-log (about 4 hours after inoculation) divided culture in half (1.5mL) and added either 37.5µL or 3.75µL of 20% arabinose solution (to try two different induction levels). Cultures grew for another hour.
- The rest of the procedure was the same as the 6/22/11 attempt but without the 42C water bath.
June 24th - Bioinformatics
Playing with Pseudocounts
Using CTC because of position 6's reliance on the CNN frequencies, we see what difference values of pseudocounts (if in the frequency table, the frequency of an amino acid is 0, bump it up to the psuedocount: ex. A = 0 becomes A = .015 with a psuedocount of .015) make. Pseudocounts are necessary for data that has small sample size - we could be missing out on working helices because a letter's frequency is 0 when it shouldn't be.
Various pseudocount (psu = ) values. Look at the 7th column, which is position 6 in the helix:
The variation from E being the top letter to A being top back to E is from a slight adjustment in how we add on psuedocounts: the 'new' way is a more proportional approach.
Notice how psu = 0 gives only the four letters found in our dataset, while psu > 0 adds in other letters, each with a small probability ranging from .5% to 2%.
The question is how much psu to add: less means we weight our (possibly flawed) data of proven zinc fingers more. Higher psu adds more randomness (variation) to our sequences, but some fraction of those sequences will not work.
Updated Closest Zif268 Fingers
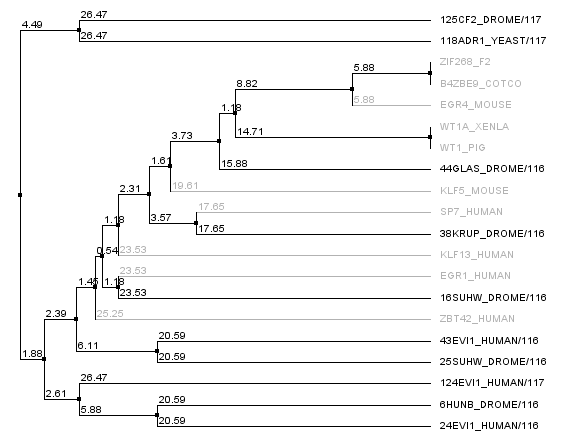
We realized that some of our "close non-zif268 fingers" were actually not all that close to Zif268, and so we went into the 88,000 zinc finger database and pulled out zinc fingers surrounding zif268. In fact, there were many, many, many zinc fingers that had identical sequences to the Zif268 F2 finger, and so we looked at sequences around it. The tree below shows the new non-zif268 backbones that are actually close to zif268 compared to our old set. The new set is in gray, the old set is in black. This gives us a potential seven more backbones to work with.
Sequence Generation
We made some small updates to the sequence generator, based on the frequencies we noticed in the outputs of the tests we ran.
- We decided to only include pseudocounts for position 6 for 'CNN' and 'ANN.' Originally, 'CNN' and 'ANN' were using pseudocounts for all seven positions. However, this introduced a noticeable increase in amino acids, such as tyrosine (Y), that have been shown to occur rarely in zinc fingers (according to our data from OPEN and Persikov). Additionally, because tryosines occured so rarely in the data (11 times total in the open data set), we decided not to give tyrosine a pseudocount.
- We added the capability to prevent repeat backbone-helix combinations on the chip. That is, we wanted to make sure that the same exact zinc finger was not generated for different triplet inputs.
To test the sequence generator, we made two sets of 2000 sequences for GAA, then infographic-d the results. Comparing these with the images for OPEN and OPEN+Persikov shows that our generation follows the major themes of those datasets, but also introduces variation. The two generated sets also vary slightly from each other, which shows the influence of randomness on the generation.
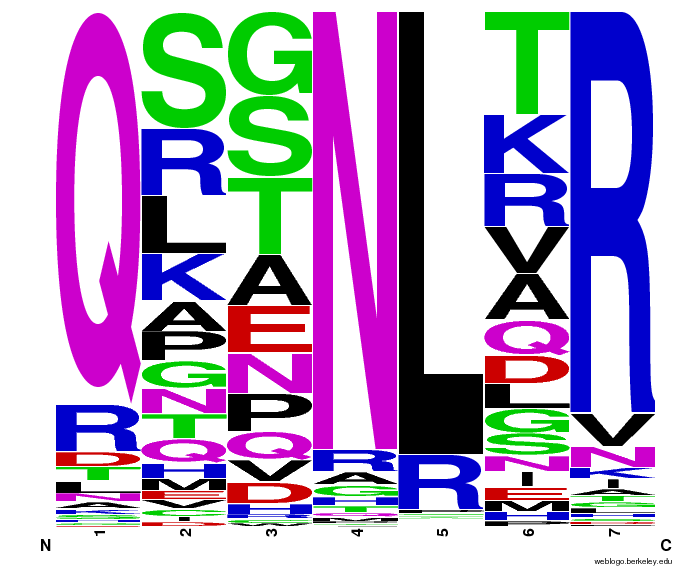 Round 1 of generating sequences for GAA with the program. |  Round 2 of generating sequences for GAA with the program. |
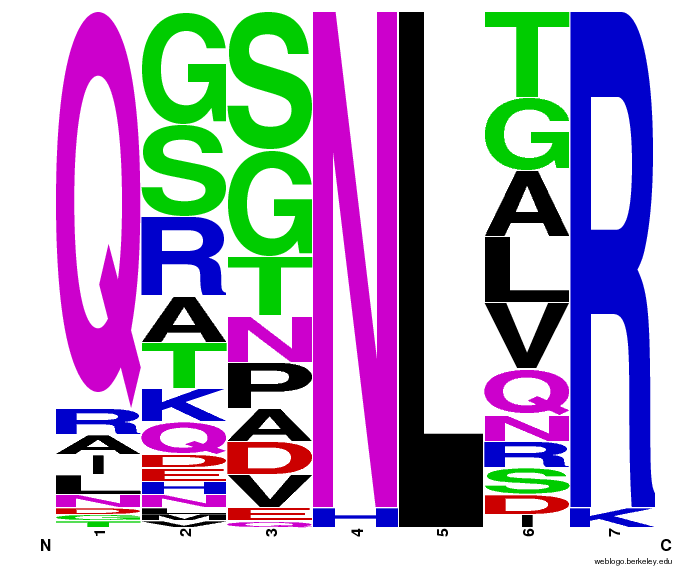 GAA sequences from the OPEN dataset. |  GAA sequences from Persikov and OPEN datasets. |
| Disease
| Target DNA Finger 1
| Helices in Zif268 Backbone
| Helices in Zif268 Closely-Related Backbones
| Helices in Zif268 Distantly-Related Backbones
|
| Colorblindness (Bottom) | TGG | 5150 | 3000 | 1000
|
| Colorblindness (Top) | ATG | 3050 | 3050 | 3050
|
| Familial Hypercholesterolemia (Bottom) | GAC | 5150 | 3000 | 1000
|
| Familial Hypercholesterolemia (Top) | CTG | 3050 | 3050 | 3050
|
| Myc (Top198) | CTC | 3050 | 3050 | 3050
|
| Myc (Top981) | AAA | 3050 | 3050 | 3050
|
Table of target sequences and helix distribution across backbones
- Distribution: Zif268 : Zif268 similar : Zif 268 dissimilar
- Conservative distribution 56.3 : 32.8 : 10.9
- Riskier distribution 33.3 : 33.3 : 33.3
List of Remaining Goals:
- Sort fingers by target
- Pick and assign primer sets
- Reverse translate fingers avoiding type II restriction enzymes and primers
- Append type II restriction enzyme and primer sequences to each finger
- Yay
June 28th
Sequencing:
- the following samples from 6/27 were sent to Genewiz for sequencing:
- PyrF F, R (one sample with PyrF_F, one with PyrF_R)
- rpoz F, R (one sample with rpoz_F, one with rpoz_R)
Lambda red results:
- the colonies on the plates did not look promising, and the ones we chose and grew up in LB+kan did not actually grow. Just to be certain, we choose 18 more colonies: 6 from 37.5µL arabinose 100µL plated, 6 from 3.75µL arabinose 100µL plated, and 6 from 37.5µL arabinose 1.5mL plated. Three from each plate were grown in plain LB and three with kan. We will let it grow in 30˚C, overnight if necessary, and hopefully see bacteria for PCR.
- Assuming this does not work, we prepared more ∆HisB∆PyrF∆rpoZ+pKD46 in two ways: we put 3 colonies in LB+amp from the 6/16 transformation plate, and we streaked a new amp plate from the glycerol stock
- Another possibility is that something is wrong with our lambda red. We designed primers to verify that the pKD46 plasmid is really in the cells.
Kan-ZFB-wp-his3-ura3 construct:
- Our last few PCR purifications have given us very low yields, and consequently we have had to use large amounts of our DNA (and the large amounts of buffer salts may also be why our lambda red recombinations have failed). When we tried to amplify our current DNA using the hisura-kan_F and ZFB-wp-hisura_R primers and the Phusion mastermix, it did not work (see 6/23). We will try to gain more product in two ways:
1) Repeat 6/23 PCR but use KAPA mastermix
- the KAPA mix may work better than the Phusion.
- Used KAPA protocol with 1µL of kan-ZFB overlap as template, hisura-kan_F and ZFB-wp-hisura_R primers, 65˚C annealing temp, 90 sec elongation time
- made 2 reactions
2) Repeat overlap extension PCR (see 6/16) with KAPA mastermix
- used KAPA protocol with 1µL kan cassette and 1µL of ZFB-wp-hisura, 65˚C annealing temp, 90 sec elongation
- 10 cycles without primers; hisura-kan_F and ZFB-wp-hisura_R primers added; 15 more cycles
- made 6 reactions
- E gel to check reactions worked: all 6 overlap PCRs successful, but not the other two reactions.
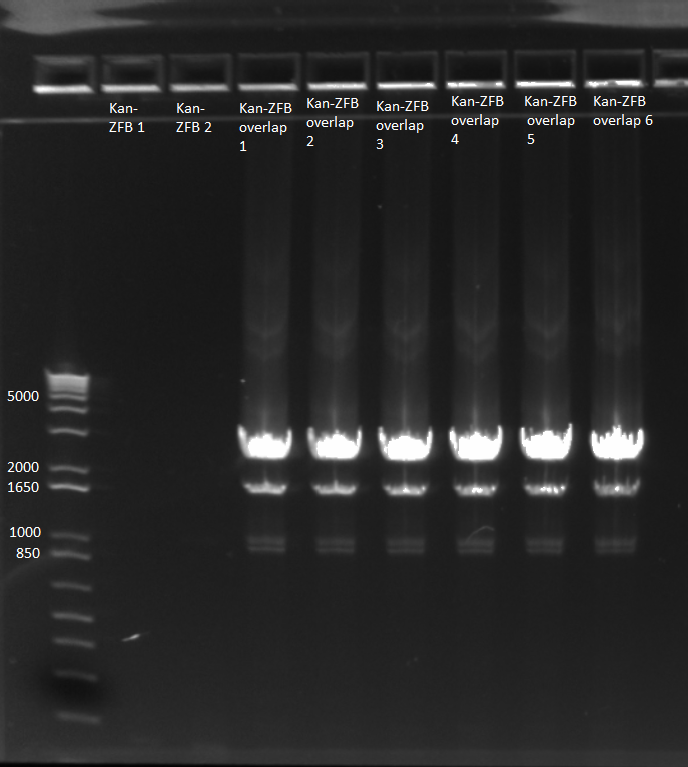
kan-ZFB-wp-hisura construct 6/28/11
- combined samples 1-3 and 4-6 and ran on 1% agarose gel for extraction
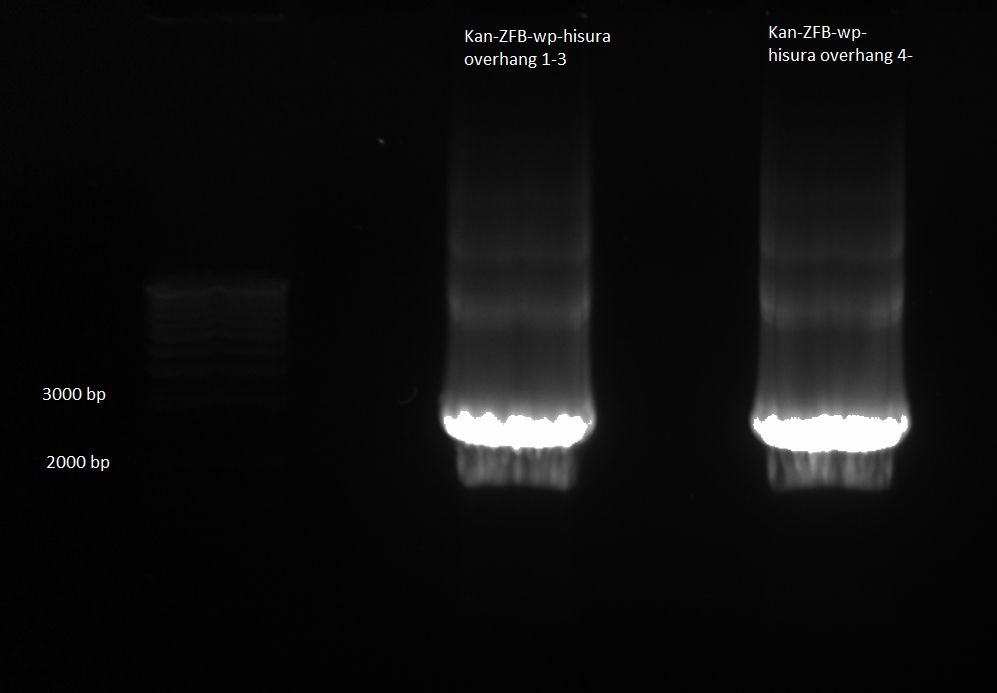
kan-ZFB-wp-hisura construct for gel extraction 6/28/11
- used Qiagen gel extraction kit and instructions with the following modifications:
- gel bands were dissolved in 500µL of buffer QG regardless of the gel volume
- gel heated at 50C for 20 min (to make up for reduced amount of buffer QG)
- after melting, 10µL of NaOAC (3M) were added to adjust the pH
- DNA from samples 1-3 were eluted in 20µL of ddH2O; DNA from samples 4-6 were eluted in 20µL of buffer EB
- water sample: 273.4 ng/µL, 260/280=1.92
- EB sample: 136.9 ng/µL, 260/280=2.38
June 28th - Bioinformatics
Attention all Harvard iGEM-ers!!! According to the iGEM Main Page, our preliminary project descriptions and safety proposals are due on July 15. Please see the aforementioned link so we can get this done ASAP- we don't want to miss any deadlines and have all our hard work wasted!
- Length of chip oligos: 131-140bp (based on Cut Site Design)
- Primers: 20bp (x2= 40bp)
- zif268 F2 backbone + helix= 23aa (x3=69bp; some fingers ~3aa longer)
- Some alternate backbones are longer than zif268 F2 backbone
- Type II binding/cut sites= 11bp on each side (22bp total)
- Standard legnth: 40 + 69 + 22 = 131bp
- Use WebLogos as a final visual check of our final generated sequences
Plasmid and Oligo Design Schematics
|
| 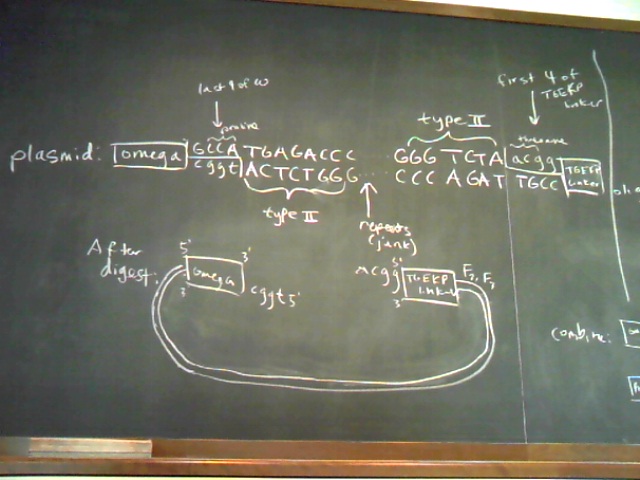 Expression Plasmid Design |
Chip-Based Sequence Design Schematic
 Chip-based process for sequence design, taken from Kosuri, et al. 2010 model of scalable gene synthesis Kosuri2010 |
References
<biblio>
- Kosuri2010 pmid=21113165
</biblio>
Harvard Logo
Running the Generator!
File:HARVFasta total.csv NOTE: LATER GENERATED NEW SEQUENCES. NOT UP TO DATE.
Generated Final Chip Sequences
- We ran the generator once earlier this afternoon, but had to re-run it again due to a typo in the cut sites and the number of sequences we desired for each backbone. Luckily, we caught these errors, and after checking the program once again, we ran it a final time this afternoon.
- It took about 45 minutes for the program to generate and reverse translate the 54900 sequences.
- During this time, we created a function that will re-translate the sequences that the generator output. It compares the original helix with the re-translated helix to make sure that our reverse-translate works properly.
- This step went smoothly, and we verified that the sequences were reverse-translated properly.
- To make sure that the distributions generated were as expected, we made WebLogos of the helices generated(see below).
- The output file (in the Dropbox: iGem > chip > final chip.csv) originally had the following headers: 'Target', 'Backbone #', 'Helix Sequence', 'Backbone Sequence', 'Nucleotide Sequence of Zinc Finger'
- We wanted to convert this information into FASTA format.
- We wrote a function that converted our original file into fasta format (in the Dropbox: iGem > chip > fasta.csv)
- The file FASTA_total (also linked above) contains the FASTA for all 50000 sequences (including the 100 controls).
- For those curious, the FASTA format just a format that looks like this:
>Header (For us the header is: Target, Backbone #, Helix Sequence, Backbone Sequence)(The header for the controls are: Index Number, 'control')
Sequence (In our case, the nucleotide sequence of the zinc fingers)
Generated WebLogos for Final Chip
- FASTA-Formatted Chip Data:
- >NNN(Target Triplet) BB# Helix Seq.
- Nucleotide seq. of ZF
Bioinformatics Candids
|
|
|  zif268 sequence by memory. You know you've stared at too many zif268 sequences when... |
File:HARVPrimer Index iGEM 2011.xls
Design of Plate Practice Sequences
While we wait for the chip to come in, we have a number of techniques and protocols that we can practice on beforehand, so that when the chip comes we'll be ready to go to use what they give us. We will be practicing the following techniques:
- Cutting ZF1 out of our oligos
- Inserting ZF1 into the expression plasmid in between the omega subunit and the linker before F2
- Verifying that combination of our F1 from the oligo with the plasmid produces a viable, functional ZF
- Amplifying subpools of oligos for testing
- Inserting the expression plasmids into the E. coli containing our selection genome
- Verifying that our ZF-binding site/GFP expression paradigm works
To this end, we will be ordering a 96-well plate from IDT containing oligos that will simulate the entire tube of oligos that we will receive from Agilent in four weeks. These oligos will consist of the following:
- 6 positive controls (we know which DNA sequences these bind to)
- 3 of them being the F1 fingers of Zif268, OZ052, and OZ123
- 3 of them being ZF F1s derived from CODA.
- 90 generated sequences, picked from a subset of the chip
- These are picked evenly across the 9,150 sequences generated on the cihp for the TGG triplet F1 target from the colorblindness "bottom finger" target, GTG GGA TGG. This particular target was chosen because the F2/F1 is a GNNTNN combo, which might be more likely to get hits from our chip generation sequences.
The primer tag sequences for the 90 generated sequence subset will be the same as they are on the chip (for the sake of explanation, we will refer to them now as P1F and P1R in this paragraph). The positive controls will be flanked immediately by the same primers as the generated subset so that we can amplify everything as one pool altogether should we need to (so this will be P1F and P1R). However, we will also put an additional set of primers outside of the P1F/P1R primers for the positive controls so that we can specifically amplify the positive control subpool, should we want to. These primers will be the same as the primers for the positive control on the chip (which will be called P2F and P2R here).
To recap, on the chip we will have the following oligos :
45750 other oligos for the 5 other target sequences
Oligos (TGG set, 9150 total): | P1F | type II binding site | generated F1 | type II binding site | P1R |
Oligos (+ control, 100 total): | P2F | type II binding site | control F1 | type II binding site | P2R |
In our test pool of 96 sequences, we will have two types of oligos (note the two pairs of primers around the positive controls):
Oligo (TGG set, 90 total): | P1F | type II binding site | generated F1 | type II binding site | P1R |
Oligo (+ control, 6 total): | P2F | P1F | type II binding site | generated F1 | type II binding site | P1R | P2R |
Once we get our test sequences back from IDT, they will come in a 96-well plate with one oligo in each plate. We should make a mixture using some of each well in order to create a tube that contains all 96 sequences. This will simulate the tube that we will receive from Agilent, except instead of 55,000 sequences we will have 96 sequences only in this tube. From here, we can practice using this as a library.
We can pretend that this tube is just 96 generated sequences on the chip, treating the positive controls as if they were also generated sequences (we only include them in the 96 to ensure that we will indeed get a "hit" from this practice screening). Thus, we can just use the P1F/P1R primer set to amplify all of them in order to use them for the subsequent steps.
These subsequent steps will be those that were outlined above, namely cutting out the F1 sequence from each oligo, ligating this F1 into our expression plasmid, putting the expression plasmid into our selection strain, observing colonies which get infused with ZFs that bind to our target site (the "hits"), and sequencing the colonies that get hits to determine which ZF they are expressing.
We will be repeating these exact same steps once we get the chip, so if we can perfect our protocols with these practice sequences, we should be golden when the chip comes in.
 "
"











.png)
.png)
.png)












.png)
.png)