|
|
| (5 intermediate revisions not shown) |
| Line 1: |
Line 1: |
| | + | <div id="723" style="display:none"> |
| | + | ==July 23rd== |
| | + | ===Team Wolfe=== |
| | + | *Lambda red results: no real colonies, only cheaters.</div> |
| | <div id="724" style="display:none"> | | <div id="724" style="display:none"> |
| | ==July 24th== | | ==July 24th== |
| Line 144: |
Line 148: |
| | ===Team ZF=== | | ===Team ZF=== |
| | ====Result of OZ052 and OZ123 Transformation==== | | ====Result of OZ052 and OZ123 Transformation==== |
| - | [[#Transformation for OZ plasmids|Last Friday]] we plated out the transformed bacteria for the two OZ controls in 10 ul and 50 ul volumes. They both yielded colonies, but the 10 ul plates only had a handful while the 50 ul plates had around 15-20 colonies each. We picked four colonies from each plate and grew them in LB/spec so that tomorrow we can perform a miniprep and send them off for sequencing. We should know the final verdict by Wednesday regarding which ones are good or bad.
| + | Last Friday we plated out the transformed bacteria for the two OZ controls in 10 ul and 50 ul volumes. They both yielded colonies, but the 10 ul plates only had a handful while the 50 ul plates had around 15-20 colonies each. We picked four colonies from each plate and grew them in LB/spec so that tomorrow we can perform a miniprep and send them off for sequencing. We should know the final verdict by Wednesday regarding which ones are good or bad. |
| | | | |
| | ====Glycerol stockmaking==== | | ====Glycerol stockmaking==== |
| - | We got the sequencing results back from last Friday, and took the following colonies to make into glycerol stock solutions: 77.1, 79.2, 80.1, 81.1 (x2 for both FH bottom and Myc 198), 82.1, 83.4, 84.1, Zif268. These colonies are a subset of the "good" colonies from our [[Lab_Notebook:_July#Sequencing_Results|sequencing results]] from Sunday. We mixed 120 ul of bacteria in mid-log with 300 ul of 80% glycerol to make the final solution, which was stored at -80 degrees C. | + | We got the sequencing results back from last Friday, and took the following colonies to make into glycerol stock solutions: 77.1, 79.2, 80.1, 81.1 (x2 for both FH bottom and Myc 198), 82.1, 83.4, 84.1, Zif268. These colonies are a subset of the "good" colonies from our sequencing results from Sunday. We mixed 120 ul of bacteria in mid-log with 300 ul of 80% glycerol to make the final solution, which was stored at -80 degrees C. |
| | | | |
| | ====Restriction enzyme test==== | | ====Restriction enzyme test==== |
| Line 160: |
Line 164: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.25.enzyme_test_1.png|thumb|E-gel of our first digestion. The gel looks unusual (notice the bubble-like formation), so we performed a second digestion.]] | + | |[[File:HARV2011.7.25.enzyme_test_1.png|thumb|E-gel of our first digestion. The gel looks unusual (notice the bubble-like formation), so we performed a second digestion.]] |
| - | |[[File:2011.7.25_enzyme_test_take2.png|thumb|1% agarose gel of our second digestion. Although the bands are faint, we can see the appropriately sized bands for Bsa1.]] | + | |[[File:HARV2011.7.25_enzyme_test_take2.png|thumb|1% agarose gel of our second digestion. Although the bands are faint, we can see the appropriately sized bands for Bsa1.]] |
| | |} | | |} |
| | | | |
| Line 173: |
Line 177: |
| | *OZ123 - 76, 126</div> | | *OZ123 - 76, 126</div> |
| | <div id="726" style="display:none"> | | <div id="726" style="display:none"> |
| | + | |
| | ==July 26== | | ==July 26== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 179: |
Line 184: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.26_ultramer_reaction_and_restriction_rerun_annotated.png|thumb|Our first PCR was extremely streaky, so we decided to redo it.]] | + | |[[File:HARV2011.7.26_ultramer_reaction_and_restriction_rerun_annotated.png|thumb|Our first PCR was extremely streaky, so we decided to redo it.]] |
| - | |[[File:2011.7.26_ultramer_touchdowns_last_four_lanes_annotated.png|thumb|We did a touchdown PCR, and our product looks better, although it's still streaky.]] | + | |[[File:HARV2011.7.26_ultramer_touchdowns_last_four_lanes_annotated.png|thumb|We did a touchdown PCR, and our product looks better, although it's still streaky.]] |
| | |} | | |} |
| | | | |
| Line 186: |
Line 191: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.26_PCR_purification_after_ultramer_assembly.jpg|thumb|Concentrations of our PCR product after PCR purification. This was used in our isothermal assembly.]] | + | |[[File:HARV2011.7.26_PCR_purification_after_ultramer_assembly.jpg|thumb|Concentrations of our PCR product after PCR purification. This was used in our isothermal assembly.]] |
| | |} | | |} |
| | | | |
| Line 209: |
Line 214: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.26_Miniprep.jpg|thumb|Our OZ concentrations after miniprep.]] | + | |[[File:HARV2011.7.26_Miniprep.jpg|thumb|Our OZ concentrations after miniprep.]] |
| | |} | | |} |
| | | | |
| | The miniprep lysis step looked good, and the samples turned blue after lysis. | | The miniprep lysis step looked good, and the samples turned blue after lysis. |
| | | | |
| - | We then performed a junction PCR using the [[Lab_Notebook:_July#PCR_of_expression_plasmid_cross-junction|previous protocol]], and checked the products on a gel: | + | We then performed a junction PCR using the previous protocol, and checked the products on a gel: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.26_oz052_and_oz123_for_seq_annotated.png|thumb|Gel of our PCR product that we sent out for sequencing.]] | + | |[[File:HARV2011.7.26_oz052_and_oz123_for_seq_annotated.png|thumb|Gel of our PCR product that we sent out for sequencing.]] |
| | |} | | |} |
| | | | |
| Line 235: |
Line 240: |
| | *Each template was analysed with presence of different sized bands for rpoZ (using rpoZ_F and rpoZ_R) and presence of Zeocin cassette (using rpoZ_R and zeocin_R) | | *Each template was analysed with presence of different sized bands for rpoZ (using rpoZ_F and rpoZ_R) and presence of Zeocin cassette (using rpoZ_R and zeocin_R) |
| | *Gel resulted with the following: | | *Gel resulted with the following: |
| - | [[File: 2011.07.26.Zeocininsertion(labeled).png|thumb|none|Zeocin insert check 7/26/11]] | + | [[File: HARV2011.07.26.Zeocininsertion(labeled).png|thumb|none|Zeocin insert check 7/26/11]] |
| | | | |
| | **'''SUCCESS!!! Yippee!!!''' | | **'''SUCCESS!!! Yippee!!!''' |
| | **NOTE. Since there was a bit of non specific annealing, we might want to use a higher annealing temperature of 64C instead of 62C | | **NOTE. Since there was a bit of non specific annealing, we might want to use a higher annealing temperature of 64C instead of 62C |
| | *Decided to run PCR for the Kan insertion just to make sure it was still present in the 4 colonies we chose, and it is!!! | | *Decided to run PCR for the Kan insertion just to make sure it was still present in the 4 colonies we chose, and it is!!! |
| - | [[File: 2011.07.26.kaninsertioncheck(labeled).png|thumb|none|Kan-ZFB-wp insert check 7/26/11]] | + | [[File: HARV2011.07.26.kaninsertioncheck(labeled).png|thumb|none|Kan-ZFB-wp insert check 7/26/11]] |
| | | | |
| | WERE READY TO ROOOCCKKK!!!!! | | WERE READY TO ROOOCCKKK!!!!! |
| Line 355: |
Line 360: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.28_practice_plate_primer_test_annotated.png|thumb|Practice plate primer test]] | + | |[[File:HARV2011.7.28_practice_plate_primer_test_annotated.png|thumb|Practice plate primer test]] |
| | |} | | |} |
| | | | |
| Line 370: |
Line 375: |
| | *PCR: colonies/culture diluted 1:20 as template (and colonies grown up simultaneously); M13_F and M13_R primers; 50˚C annealing and 3min elongation; 25 cycles | | *PCR: colonies/culture diluted 1:20 as template (and colonies grown up simultaneously); M13_F and M13_R primers; 50˚C annealing and 3min elongation; 25 cycles |
| | *Results: bands are similar to the ones seen the last time we did this PCR (and those samples had also been sequence-verified). These colonies should be good to go! | | *Results: bands are similar to the ones seen the last time we did this PCR (and those samples had also been sequence-verified). These colonies should be good to go! |
| - | [[File: 2011.07.29.pSR01check(labeled).png|thumb|none|pSR01 check 7/29/11]] | + | [[File: HARV2011.07.29.pSR01check(labeled).png|thumb|none|pSR01 check 7/29/11]] |
| | '''pSB4K5:''' | | '''pSB4K5:''' |
| | *Miniprepped both cultures. Yields weren't great (in the case of 2:17H, there actually was a spill) but they are still usable. | | *Miniprepped both cultures. Yields weren't great (in the case of 2:17H, there actually was a spill) but they are still usable. |
| Line 380: |
Line 385: |
| | **58˚C anneal, 3 min elongation, 25 cycles | | **58˚C anneal, 3 min elongation, 25 cycles |
| | *Results: the product was really really really faint--primer dimers were by far the bulk of the product. We realized this is probably because one of the primers includes a Not1 restriction site, which is palindromic, 8 bases long, and all Cs and Gs. | | *Results: the product was really really really faint--primer dimers were by far the bulk of the product. We realized this is probably because one of the primers includes a Not1 restriction site, which is palindromic, 8 bases long, and all Cs and Gs. |
| - | [[File: 2011.07.29.pSB4K5(labeled).png|thumb|none|pSB4K5 PCR to add homology 7/29/11]] | + | [[File: HARV2011.07.29.pSB4K5(labeled).png|thumb|none|pSB4K5 PCR to add homology 7/29/11]] |
| | *In order to counteract the primer dimers, we will try a gradient PCR with a range of higher annealing temperatures: | | *In order to counteract the primer dimers, we will try a gradient PCR with a range of higher annealing temperatures: |
| | **same protocol as before except the annealing temp went from 58-65 with 8 samples (1=58, 8=65) | | **same protocol as before except the annealing temp went from 58-65 with 8 samples (1=58, 8=65) |
| | **Results: 3:11P did have visible product bands, but the primer dimers are still stronger | | **Results: 3:11P did have visible product bands, but the primer dimers are still stronger |
| - | [[File: 2011.07.29 pSB4K5 grad(labeled).png|thumb|none|pSB4K5 gradient PCR 7/29/11]] | + | [[File: HARV2011.07.29 pSB4K5 grad(labeled).png|thumb|none|pSB4K5 gradient PCR 7/29/11]] |
| | | | |
| | ===Team TolC=== | | ===Team TolC=== |
| Line 393: |
Line 398: |
| | *So we chose 5 colonies and ran PCR on them to gel them out | | *So we chose 5 colonies and ran PCR on them to gel them out |
| | **The gel shows success for colonies 2,3,4, and 5 that we chose!!! | | **The gel shows success for colonies 2,3,4, and 5 that we chose!!! |
| - | [[File: 2011.07.29.zif268plasmidcheck1.png|thumb|none|Zif268 plasmid insertion check 7/29/11]] | + | [[File: HARV2011.07.29.zif268plasmidcheck1.png|thumb|none|Zif268 plasmid insertion check 7/29/11]] |
| | | | |
| | '''READY FOR SELECTION!!!''' | | '''READY FOR SELECTION!!!''' |
| Line 399: |
Line 404: |
| | ===Team ZF=== | | ===Team ZF=== |
| | ====Transformation plating results==== | | ====Transformation plating results==== |
| - | We got no growth on any of the plated transformed bacteria for either the ElectroComp or ChemComp bacteria. To check whether the isothermal assembly itself was the problem, we ran a junction PCR using our [[Lab_Notebook:_July#PCR_of_expression_plasmid_cross-junction|previous protocol]] on the isothermal assembly mix, along with a positive control (Myc981): | + | We got no growth on any of the plated transformed bacteria for either the ElectroComp or ChemComp bacteria. To check whether the isothermal assembly itself was the problem, we ran a junction PCR using our previous protocol on the isothermal assembly mix, along with a positive control (Myc981): |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.29_isothermal_assembly_junction_test_annotated.png|thumb|No bands visible for the isothermal assembly]] | + | |[[File:HARV2011.7.29_isothermal_assembly_junction_test_annotated.png|thumb|No bands visible for the isothermal assembly]] |
| | |} | | |} |
| | | | |
| Line 431: |
Line 436: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.29_Practice_Plate_oligo_pcr_pure.jpg|thumb|DNA concentrations]] | + | |[[File:HARV2011.7.29_Practice_Plate_oligo_pcr_pure.jpg|thumb|DNA concentrations]] |
| | |} | | |} |
| | | | |
| Line 440: |
Line 445: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.29_practice_plate_neg_controls_annotated.png|thumb|Strange results]] | + | |[[File:HARV2011.7.29_practice_plate_neg_controls_annotated.png|thumb|Strange results]] |
| | |} | | |} |
| | | | |
| Line 453: |
Line 458: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.30_CB_bottom_miniprep.jpg|thumb|DNA concentrations after miniprep]] | + | |[[File:HARV2011.7.30_CB_bottom_miniprep.jpg|thumb|DNA concentrations after miniprep]] |
| | |} | | |} |
| | | | |
| Line 466: |
Line 471: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.7.31_primer_tag_test_two_annotated.png|thumb|PCR repeat results]] | + | |[[File:HARV2011.7.31_primer_tag_test_two_annotated.png|thumb|PCR repeat results]] |
| | |} | | |} |
| | | | |
July 25th
Team TolC
Sequencing of Jun's rpoZ MAGE knockout
- Prepared six products and primers for sequencing
- Sending Jun colony 4, PCR's 1-mutant_R, 2-mutant_R, 3-mutant_R, 4-mutant_R, 2-mutant_Int_R, and 4-mutant_Int_R
- delayed until tomorrow (July 25, 2011) because pickup for the day already done.
Lambda Red Recombination of zeocine into TolC(assistance from Naomi, Cub team member)
- PCR out zeocin cassette (replacing the rpoZ, for selection) from Wolfe Strain
- Used primers rpoZ_F and rpoz_R, expecting a band about 1500bp? Total length about 939bp, including the promoters, according to http://rothlab.ucdavis.edu/drugs/zeo.html Roth lab @ UC Davis, Zeocin
- Ran 8, 25 µL solutions with KAPA
- PCR protocol(PCR 7 machine, "ZEOC")
- 95˚ C, for 5 min
- 98˚ C, for 20 sec
- 62˚ C, for 15 sec
- 72˚ C, for 45 sec
- Repeat 29 times
- 72˚ C, for 5 min
- 4˚ C, forever
- Gel extracted the PCR product NOTE. Since the gel, was so clean, we may do a PCR clean up next time, instead of running the PCR product through a gel for gel extraction. Just make sure to use these exact same parameters.
- Nanodrop
- 156.2 ng/µL and 107.6 ng/µL, each in 30ul of water (distilled, deionised- molecular grade)
- Performed lambda red recombination
rpoZ MAGE knockout attempt...yet again
- Used new oligo to perform a MAGE knockout of rpoZ
- Oligo named rpoZ_MAGE_Subs
- Electroporator read 5.2 ms
- Allowed for 3 hours of growth and spread on zeocine plate
Team Wolfe Cub
- Plate reader is still being repaired--we will have to wait to check the growth curves
- Since plans A and B are not working well at the moment, we will go ahead and start preparing for plan C (using a plasmid). Ordered primers that will clone out the pSB4K5 (a biobrick vector with a kan cassette, http://partsregistry.org/Part:pSB4K5) backbone with homology to the ZFB-wp-his3-ura3 construct. We will then connect the two with Gibson assembly.
- To this end, we need to use the biobrick DNA sent to us to transform chemically competent E coli. Unfortunately the 384-well plates that they are stored in could not be found because Andrew is out of town--hopefully they will turn up tomorrow.
- For the rest of the day, we joined team TolC for gel extraction, MAGE, and lambda red!
Team ZF
Result of OZ052 and OZ123 Transformation
Last Friday we plated out the transformed bacteria for the two OZ controls in 10 ul and 50 ul volumes. They both yielded colonies, but the 10 ul plates only had a handful while the 50 ul plates had around 15-20 colonies each. We picked four colonies from each plate and grew them in LB/spec so that tomorrow we can perform a miniprep and send them off for sequencing. We should know the final verdict by Wednesday regarding which ones are good or bad.
Glycerol stockmaking
We got the sequencing results back from last Friday, and took the following colonies to make into glycerol stock solutions: 77.1, 79.2, 80.1, 81.1 (x2 for both FH bottom and Myc 198), 82.1, 83.4, 84.1, Zif268. These colonies are a subset of the "good" colonies from our sequencing results from Sunday. We mixed 120 ul of bacteria in mid-log with 300 ul of 80% glycerol to make the final solution, which was stored at -80 degrees C.
Restriction enzyme test
We tested both XbaI and BsaI on the junction PCR product using colony 79.4 as a template (which was used because it was a "bad" sequence overall but had intact BsaI and XbaI cut sites). The junction PCR product is 1326 bp, and we expected to see bands of 593 and 739 bp with XbaI, and 717 and 578 bp with BsaI. Our restriction digest recipe consisted of the following per reaction:
- 1 ul 10X buffer (NE biolabs buffer 4)
- 7.9 ul H2O
- 1 ul DNA (~100 ng)
- 1 ul enzyme (~2 units)
- 1 ul BSA (100x)
The two gels we ran for this digest showed that XbaI did NOT work, which may be due to XbaI being too old. However, we did get double bands both times for BsaI. Our egel was strange because of uneven loading, and so we were unable to determine the true size of both bands, but the agarose gel we ran later showed the two bands in the right positions. The two gels can be seen below:
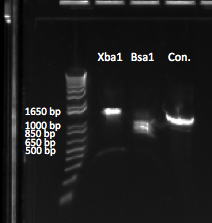 E-gel of our first digestion. The gel looks unusual (notice the bubble-like formation), so we performed a second digestion. |  1% agarose gel of our second digestion. Although the bands are faint, we can see the appropriately sized bands for Bsa1. |
Ultramer Construction of Pos Con 77 and Other F2/F3 positive controls
We redid the ultramer overlap PCR for Pos Con 77, this time with the correct reverse sequence. We also assembled ultramers for Zif268, OZ052 and OZ123 (these are the plasmids that will have the type II cut sites in them along with F2/F3 only, as opposed to having the entire finger already assembled). For the Zif268 reaction, an annealing temperature of 65*C was used, and for the rest, the annealing temperature was 58*C. For all reactions, the extension time was 15 seconds.
The following primers were used for each reaction (index taken from the Dropbox primer index):
- Pos con 77 - 76, 122
- Zif268 - 123, 124
- OZ052 - 76, 125
- OZ123 - 76, 126
July 26
Team ZF
Isothermal Assembly of Pos Con 77, Zif 268, and OZs
Today we checked the PCR products of the ultramer reaction from yesterday. They did not look good. We repeated the reaction using a touchdown PCR beginning with an annealing temperature of 70 degrees and decreasing 2 degrees every other cycle until hitting a minimum of 58 degrees, and we brought the total number of cycles to 25. Extension time was 30 seconds. This looked great. The pictures for the two reactions can be seen below:
 Our first PCR was extremely streaky, so we decided to redo it. |  We did a touchdown PCR, and our product looks better, although it's still streaky. |
Before the isothermal assembly, we did a PCR purification and measured the concentrations of the ultramer reaction products:
 Concentrations of our PCR product after PCR purification. This was used in our isothermal assembly. |
We then performed an isothermal assembly, using the following mix for Pos con 77, OZ052, and OZ123:
- 0.63 ul of spec backbone at ~157 ng/ul
- 0.3 ul of ultramer (~50 ng/ul)
- 0.13 ul of omega subunit/linker
- 1.44 ul of H2O
- 7.5 ul of isothermal mix
For Zif268:
- 0.63 ul of spec backbone
- 0.19 ul of Zif (~90 ng/ul)
- 0.13 ul of omega/linker
- 1.55 of H2O
- 7.5 ul of isothermal mix
This was left at 50 degrees for an hour, then stored at 4 degrees.
Prepare control OZs for sequencing
We performed a miniprep on the eight samples for sequencing, obtaining the following concentrations:
 Our OZ concentrations after miniprep. |
The miniprep lysis step looked good, and the samples turned blue after lysis.
We then performed a junction PCR using the previous protocol, and checked the products on a gel:
 Gel of our PCR product that we sent out for sequencing. |
They looked good, so we sent them out for sequencing. Hopefully we will have our final OZ control plasmids for tomorrow.
Practice Plate resuspension
We resuspended the practice plate oligos in 40 ul of water to produce a 100 uM solution, taking 10 ul of the final solutions into 90 ul of water in separate tubes to create 10 uM working concentrations. When we receive our tag primers, we should be able to test the presence of the oligos in each tube as well as amplify them in preparation for integration into the expression plasmids.
Colony finalization
For the colonies that we plated yesterday, we saw that the plates all had good growth on them and so we wrapped the plates in parafilm and put them at 4 degrees for storage. 1/1000 ul from saturated culture seems to be a good amount to plate.
Team TolC
Zeocin insertion into ECNR2
- Plated 3 mL of our recombination culture on zeocin plate, and also plated 2 mL of MAGE results on zeocin plate as a control
- The recombination plate had about 15 colonies around 9 30am, and the MAGE plate had none
- Continued by PCR of 4 colonies on recombination plate
- Each template was analysed with presence of different sized bands for rpoZ (using rpoZ_F and rpoZ_R) and presence of Zeocin cassette (using rpoZ_R and zeocin_R)
- Gel resulted with the following:

Zeocin insert check 7/26/11
- SUCCESS!!! Yippee!!!
- NOTE. Since there was a bit of non specific annealing, we might want to use a higher annealing temperature of 64C instead of 62C
- Decided to run PCR for the Kan insertion just to make sure it was still present in the 4 colonies we chose, and it is!!!

Kan-ZFB-wp insert check 7/26/11
WERE READY TO ROOOCCKKK!!!!!
MAGE rpoZ knockout
- Plated 1:10 and 1:100 dilution of MAGE results, and this morning at 10 am, they both had cells and in appropriate proportions
- If above PCR and gel of zeocin insertion does not appear to be successful, then we will run 50 PCR on 50 different colonies on the 1:100 dilution plate of MAGE rpoZ knockout
Team Wolfe
- Plate reader: trying to get in contact with Christian Daly who runs the Spectramax plate reader in the Bauer facility (Northwest basement). Hopefully we will get access soon.
- Selection system plasmid: still trying to get hold of the biobricks. Designed Quikchange primers to change the zinc finger binding sites.
- Lambda red recombination: gave lambda red one last chance, this time with a positive control
- negative control (water): plated 1mL
- positive control (kan cassette from Wyss): 50ng DNA, plated 1mL and 100uL
- thio kan-ZFB-wp-his3-ura3: 180ng DNA, plated 1mL
July 28th
Team Wolfe
Plate reader and pSR01:
- the Bauer plate reader seems to have worked very well, bu† unfortunately none of our pSR01-transformed colonies grew. This is probably because the plates were a couple weeks old. Unfortunately our glycerol stock of the strain also is missing, so we will try growing up some of the colonies in case on of them works. Otherwise, we will grow up ∆HisB∆pyrF∆rpoZ and transform again.
- Transformation: usual protocol, about 100µL DNA, recovery 2hrs, plate 1µL, 10µL, 100µL.
pSB4K5:
- The kan plates from the transformation all had colonies! We chose one from 2:17H and one from 3:11P to grow up in LB+kan for miniprep and glycerol stocks.
- Ordered quikchange primers: for OZ052 and OZ123 we added the context bases 5' and 3' of the binding sequence (found in OPEN supplementary materials). Since CODA did not include the extra bases, we left them the same as the original plasmid (i.e. Zif268's).
Team TolC
cultures of ECNR2+Kan-ZFB-wp+zeocin from F9,F11, for glycerol stocks and transformation
Sequencing
- Ran PCR and sent out sequencing to solidify the presence of the Kan+ZFB+wp and zeocin within the ECNR2 genome
- Used rpoZ_R, rpoZ_F for PCR of the zeocin cassette (and rpoZ_F for sequencing) and TolC_Seq_R, TolC_Seq_F for PCR of the Kan-ZFB-wp insert (and tolC_seq_F for sequencing)
Plasmid transformation
- Performed transformation on the Zif268 plasmid into the ECNR2 culture
- Used 3 µL in a 50 µL dilution (49+1) of 1 ul of 55 ng/µL concentration of plasmids
- 1.82 V for 5.00 ms according to the electroporator
Team ZF
Checking on Pos Con 77, OZs, and Zif 268 plates
Unfortunately, nothing grew on our plates from yesterday. Our time constants from the electroporation were rather low (~3.8), so 50 ul may not be enough to see growth. We decided to spin down the cells, resuspend them in 50 ul of LB, and re-plated this solution. We also re-did the transformation using ChemComp cells, and made 100 ul plates. Both sets of plates were left overnight at 37*.
Designing the reporter strain
Instead of placing the GFP reporter on a separate plasmid, we decided to insert it on our expression plasmid, immediately after our zinc fingers, and before the transcription terminator. We placed a ribosome binding site (Shine Dalgarno sequence: AGGAGGTT before the GFP. We will only add the GFP to Zif 268 (whole) because if we can show through GFP that the selection system works for Zif 268, we can assume it also works for the other zinc fingers. This is easier and more cost effective than ordering 12 different primers for each of our plasmids.
We will construct the reporter strain by performing a PCR to get the plasmid in linear form. We will also perform a second PCR to get out GFP and add homology regions to our Zif 268 plasmid, as well as adding the Shine-Dalgarno sequence. The primers for these PCRs were ordered today. We'll then assemble these pieces to construct a whole plasmid, and transform our bacteria.
PCR with Practice Plate
We performed a PCR to amplify our F1s from the practice plate. We used our primers for CB bottom (primer index: 66 and 67) for the 18 targets and our primers for the controls (primer index: 72 and 73) for the 6 controls. We had an annealing temperature of 52 degrees and an extension time of 15 seconds.
 Practice plate primer test |
We got products for all of the oligos at our expected product size of 131 bp.
Glycerol stocks of successful plasmids
We happened to check our glycerol stocks, and noticed that the glycerol and bacteria were not mixed thoroughly. We remade all of our glycerol stocks and put them into the -80*.
July 29th
Team Wolfe
pSR01:
- The colonies in the 96 well plate did grow overnight, but they were not saturated, implying that they were very unhealthy. We will PCR a few of them (1,2,3,6) just to see if they actually have the plasmid still
- Transformation plates look great--we will PCR 12 colonies to verify the plasmid is present
- PCR: colonies/culture diluted 1:20 as template (and colonies grown up simultaneously); M13_F and M13_R primers; 50˚C annealing and 3min elongation; 25 cycles
- Results: bands are similar to the ones seen the last time we did this PCR (and those samples had also been sequence-verified). These colonies should be good to go!
pSB4K5:
- Miniprepped both cultures. Yields weren't great (in the case of 2:17H, there actually was a spill) but they are still usable.
- 2:17H: 6.4 ng/µL 260/280=2.2
- 3:11P: 9.6ng/µL 260/280=2.4
- PCR to add ZFB-wp-his3-ura3 homology:
- 1µL of plasmid as template
- pSB4K5_F+ZFBhl and pSB4K5_R+URA3hl primers
- 58˚C anneal, 3 min elongation, 25 cycles
- Results: the product was really really really faint--primer dimers were by far the bulk of the product. We realized this is probably because one of the primers includes a Not1 restriction site, which is palindromic, 8 bases long, and all Cs and Gs.
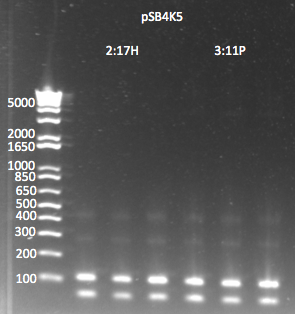
pSB4K5 PCR to add homology 7/29/11
- In order to counteract the primer dimers, we will try a gradient PCR with a range of higher annealing temperatures:
- same protocol as before except the annealing temp went from 58-65 with 8 samples (1=58, 8=65)
- Results: 3:11P did have visible product bands, but the primer dimers are still stronger

pSB4K5 gradient PCR 7/29/11
Team TolC
Sequencing of Kan+ZFB+wp and Zeocin insertions
- Sequencing results were successful and solidified presence of both insertions
Zif268 Plasmid Insertion into ECNR2+Kan-ZFB-wp+zeocin
- The plates had growth on them!!!
- So we chose 5 colonies and ran PCR on them to gel them out
- The gel shows success for colonies 2,3,4, and 5 that we chose!!!

Zif268 plasmid insertion check 7/29/11
READY FOR SELECTION!!!
Team ZF
Transformation plating results
We got no growth on any of the plated transformed bacteria for either the ElectroComp or ChemComp bacteria. To check whether the isothermal assembly itself was the problem, we ran a junction PCR using our previous protocol on the isothermal assembly mix, along with a positive control (Myc981):
 No bands visible for the isothermal assembly |
As observed, there were no bands in the isothermal mix lanes while the positive control worked. Thus, we can conclude that our isothermal assembly did not work, and we will need to repeat it again (currently waiting for more isothermal mix to arrive from Wyss).
Amplification and purification of practice plate oligos
In preparation for our swap reactions (switching the oligo F1s into our expression plasmid), we mapped out the following two experiments that we will need to perform:
- Testing the selection system by preparing our expression plasmids in parallel (i.e. creating 18 plasmids in separate reactions), and then combining them all at the end and transforming them into the selection strains. This will allow us to cut and ligate the plasmids without much concern, but test whether we will be able to pick out the binder amongst the rest of the non-binders.
- Testing the cutting/ligation by combining all of the oligos first, and then carrying out the construction of the different expression plasmids all in the same tube.
Note that the results for both will ultimately yield DNA libraries if they work (except that the first experiment is much more foolproof in terms of actually yielding a library than the second experiment).
To carry out the first experiment, the following steps need to be undertaken:
- Perform PCRs on each of the practice plate oligos such that we end up with a relatively high concentration of DNA after PCR and PCR purification (over 40 ng/ul).
- Cut both plasmid backbone and oligos with BsaI type II restriction enzyme.
- PCR purify restriction products to get rid of enzyme and junk DNA.
- Run phosphatase reaction to prevent autoligation.
- Mix and ligate to get full plasmid.
- Some tips:
- Each ligation reaction should only have 10-15 ul volume.
- You should have 100 ng of backbone per reaction
- Since we are planning 18 reactions, we will need 1800 ng of backbone, which is several minipreps worth
- The backbone:oligo ratio should be appoximately 1:4
- In our case, the backbone is 2.2 kb and the insert is 131 bp, so for 100 ng of backbone there should be ~20 ng of insert
- Gel extract plasmid to get final library.
Today, we did PCRs and PCR purifications for step one as outlined above. We tested the concentrations of the results, displayed below:
In order to have enough backbone to work with, we seeded some LB/spec with CB bottom bacteria in preparation for 4 minipreps of 3 mL each tomorrow. This should definitely yield the 1800 ng of backbone we will need for the ligation.
Practice plate primer tag test
We tested the primers to see if their selection of subpools was exclusive enough or not (i.e. to make sure that our control primers did not amplify the CB bottom sequences, etc.). We ran a PCR crossing primers with DNA with the following results:
The first and third lanes are CB primer/CB DNA and control primer/control DNA respectively, which should yield bands. Lanes two and four were control primer/CB DNA and CB primer/control DNA respectively, which should yield no bands. The fact that we got bands for these lanes is a little bit worrying, and so we are repeating this PCR again to see if we get the same results when we run the gel tomorrow.
 "
"







.png)
.png)

.png)
.png)
.png)





