|
|
| (8 intermediate revisions not shown) |
| Line 1: |
Line 1: |
| - | <div style="display:none"> | + | <div id='805' style="display:none"> |
| | ==August 5th== | | ==August 5th== |
| | ===Team Wolfe=== | | ===Team Wolfe=== |
| Line 31: |
Line 31: |
| | After talking with Dan about the not-so-ideal experiences with Bsa-HF, we decided to make a few changes to our protocols, and that we would try the following to see how they would work: | | After talking with Dan about the not-so-ideal experiences with Bsa-HF, we decided to make a few changes to our protocols, and that we would try the following to see how they would work: |
| | | | |
| - | ''To troubleshoot the [[Lab_Notebook:_August#Practice_plate_ligation|poor oligo cutting]]:''<br /> | + | ''To troubleshoot the poor oligo cutting:''<br /> |
| | 1.5 ul of enzyme (20 units/ul)<br /> | | 1.5 ul of enzyme (20 units/ul)<br /> |
| | 2 ul of NEB buffer 4<br /> | | 2 ul of NEB buffer 4<br /> |
| Line 42: |
Line 42: |
| | Denature at 65 degrees for 20 minutes and then pcr purify. | | Denature at 65 degrees for 20 minutes and then pcr purify. |
| | | | |
| - | ''For troubleshooting the [[Lab_Notebook:_August#Ligation_of_oligos_and_backbone|poor plasmid cutting]]:''<br /> | + | ''For troubleshooting the poor plasmid cutting:''<br /> |
| | Do the same thing but do it for longer (2 hours) and also run an uncut plasmid to see which bands come up. | | Do the same thing but do it for longer (2 hours) and also run an uncut plasmid to see which bands come up. |
| | | | |
| Line 51: |
Line 51: |
| | | | |
| | ====Glycerol stocks and final platings==== | | ====Glycerol stocks and final platings==== |
| - | We made glycerol stocks of the final four plasmids in Top 10 ChemComp bacteria, which were Pos Con 77 (85.2), OZ052 (52.1), OZ123 (123.4), and Zif268 (Zif268.1). We also made final plates of these bacteria, using 1/1000 ul on spec plates. | + | We made glycerol stocks of the final four plasmids in Top 10 ChemComp bacteria, which were Pos Con 77 (85.2), OZ052 (52.1), OZ123 (123.4), and Zif268 (Zif268.1). We also made final plates of these bacteria, using 1/1000 ul on spec plates.</div> |
| - | | + | <div id='806' style="display:none"> |
| | ==August 6th== | | ==August 6th== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 74: |
Line 74: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.6_manual_miniprep_cb_bottom_awesome.jpg|thumb|Over 20x times normal yield compared with the Qiagen miniprep kit.]] | + | |[[File:HARV2011.8.6_manual_miniprep_cb_bottom_awesome.jpg|thumb|Over 20x times normal yield compared with the Qiagen miniprep kit.]] |
| | |} | | |} |
| | | | |
| Line 101: |
Line 101: |
| | *HisB bands at about 900bp; PyrF bands at about 400bp. | | *HisB bands at about 900bp; PyrF bands at about 400bp. |
| | *Results: none of the colonies have both genes knocked out but some do look like they may have the HisB deletion. We may try again using MAGE round 5 colonies. | | *Results: none of the colonies have both genes knocked out but some do look like they may have the HisB deletion. We may try again using MAGE round 5 colonies. |
| - | [[File: 2011.08.06.hispyrKOtest1(labeled).png|thumb|left|HisB and PyrF allele specific PCRs part 1. Yellow-labeled colonies may have a knock out. 8/6/11]] | + | [[File:HARV2011.08.06.hispyrKOtest1(labeled)-0016.png|thumb|left|HisB and PyrF allele specific PCRs part 1. Yellow-labeled colonies may have a knock out. 8/6/11]] |
| - | [[File: 2011.08.06.pyrfKOtest(labeled).png|thumb|none|Separate E gel of the pyrF PCRs cut off in gel 1. 8/6/11]] | + | [[File:HARV2011.08.06.pyrfKOtest(labeled)-0018.png|thumb|none|Separate E gel of the pyrF PCRs cut off in gel 1. 8/6/11]] |
| - | [[File: 2011.08.06.hispyrKOtest2_2(labeled).png|thumb|none| HisB and PyrF allele specific PCRs part 2 8/6/11]] | + | [[File:HARV2011.08.06.hispyrKOtest2_2(labeled)-0017.png|thumb|none| HisB and PyrF allele specific PCRs part 2 8/6/11]] |
| | '''pSB4K5 digestion:''' | | '''pSB4K5 digestion:''' |
| | *Since our minipreps have been giving such low yields, we tried an [http://openwetware.org/wiki/Knight:Miniprep_low_copy_plasmids altered protocol]. The result, however was the same: 6mL of culture produced 19ng/uL (in 30uL) | | *Since our minipreps have been giving such low yields, we tried an [http://openwetware.org/wiki/Knight:Miniprep_low_copy_plasmids altered protocol]. The result, however was the same: 6mL of culture produced 19ng/uL (in 30uL) |
| Line 113: |
Line 113: |
| | **.72uL BSA | | **.72uL BSA |
| | **3 hour incubation at 37C | | **3 hour incubation at 37C |
| - | *gel extraction: the gel was empty. No clue why. Obviously this is a setback--we will have to redo the miniprep and digestion before we can form the whole plasmid | + | *gel extraction: the gel was empty. No clue why. Obviously this is a setback--we will have to redo the miniprep and digestion before we can form the whole plasmid.</div> |
| - | | + | <div id='807' style="display:none"> |
| | ==August 7th== | | ==August 7th== |
| | ===Team ZF=== | | ===Team ZF=== |
| Line 123: |
Line 123: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.7_epic_miniprep.png|thumb|Hopefully this is actually real, it seems almost too good to be true.]] | + | |[[File:HARV2011.8.7_epic_miniprep.png|thumb|Hopefully this is actually real, it seems almost too good to be true.]] |
| | |} | | |} |
| | | | |
| Line 134: |
Line 134: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.7_oligo_cut_and_plasmid_test_annotated.png|thumb|blah]] | + | |[[File:HARV2011.8.7_oligo_cut_and_plasmid_test_annotated.png|thumb|blah]] |
| | |} | | |} |
| | | | |
| Line 144: |
Line 144: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.7_CB_bottom_cut_plasmid_annotated.png|thumb|blah]] | + | |[[File:HARV2011.8.7_CB_bottom_cut_plasmid_annotated.png|thumb|blah]] |
| | |} | | |} |
| | | | |
| - | Although the bands are faint, the cut band is somewhere around the correct size (around 2.8 kb). Although the band appears to be anywhere between 3 kb and 4 kb in the gel, the ladder ran strangely for some reason. However, there is still a fairly well-defined band instead of a smear in the cut lane. This in itself is a huge relief - if there were significant genomic DNA contamination, we might expect to get a smear from many differently sized cuts. Furthermore, the uncut band looks standard for a plasmid, similar to the picture on the website here: [[http://faculty.plattsburgh.edu/donald.slish/Electrophoresis.html]]. There are three bands in our gel as they appear on the website. However, our linearized plasmid is actually fairly close to the bottom band for the uncut plasmid, which is presumably the supercoiled form. | + | Although the bands are faint, the cut band is somewhere around the correct size (around 2.8 kb). Although the band appears to be anywhere between 3 kb and 4 kb in the gel, the ladder ran strangely for some reason. However, there is still a fairly well-defined band instead of a smear in the cut lane. This in itself is a huge relief - if there were significant genomic DNA contamination, we might expect to get a smear from many differently sized cuts. Furthermore, the uncut band looks standard for a plasmid, similar to the picture on the website here: [[http://faculty.plattsburgh.edu/donald.slish/Electrophoresis.html]]. There are three bands in our gel as they appear on the website. However, our linearized plasmid is actually fairly close to the bottom band for the uncut plasmid, which is presumably the supercoiled form.</div> |
| - | | + | <div id='808' style="display:none"> |
| | ==August 8th== | | ==August 8th== |
| | ===Team Wolfe=== | | ===Team Wolfe=== |
| Line 163: |
Line 163: |
| | *64 degrees annealing and 90 seconds elongation | | *64 degrees annealing and 90 seconds elongation |
| | *Gel shows PCR was successful | | *Gel shows PCR was successful |
| - | [[File: 2011.08.08 His Pyr MAGE for Seq (labeled).png|thumb|none|MAGE sequencing PCR]] | + | [[File: HARV2011.08.08 His Pyr MAGE for Seq (labeled)-0019.png|thumb|none|MAGE sequencing PCR]] |
| | | | |
| | | | |
| Line 195: |
Line 195: |
| | We were able to access the results of our GFP sequencing. | | We were able to access the results of our GFP sequencing. |
| | | | |
| - | {| {{table}} | + | {| |
| | | align="center" style="background:#f0f0f0;"|''' ''' | | | align="center" style="background:#f0f0f0;"|''' ''' |
| | | align="center" style="background:#f0f0f0;"|'''GFP Forward''' | | | align="center" style="background:#f0f0f0;"|'''GFP Forward''' |
| Line 231: |
Line 231: |
| | | | |
| | ====Re-do minipreps for control backbones with new protocol==== | | ====Re-do minipreps for control backbones with new protocol==== |
| - | We used the [[#New miniprep protocol|new protocol]] to do a miniprep on the control backbones. | + | We used the new protocol to do a miniprep on the control backbones. |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.8_control_backbone_miniprep.jpg|thumb|We got (unusually?) high yields using the new technique.]] | + | |[[File:HARV2011.8.8_control_backbone_miniprep.jpg|thumb|We got (unusually?) high yields using the new technique.]] |
| | |} | | |} |
| | | | |
| Line 240: |
Line 240: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.8_miniprep_cut_controls_annotated.png|thumb|We ran a gel of our new miniprep product.]] | + | |[[File:HARV2011.8.8_miniprep_cut_controls_annotated.png|thumb|We ran a gel of our new miniprep product.]] |
| | |} | | |} |
| | | | |
| Line 246: |
Line 246: |
| | | | |
| | ====Ligation and transformation of Oligo 5 and 6 and CB Bottom==== | | ====Ligation and transformation of Oligo 5 and 6 and CB Bottom==== |
| - | We decided to try the ligation of our oligos and the backbone, using our [[Lab_Notebook:_August#Practice_plate_ligation|previous protocol]]. We then transformed the product using ChemComp cells, plating 50 and 450 ul. | + | We decided to try the ligation of our oligos and the backbone, using our previous protocol. We then transformed the product using ChemComp cells, plating 50 and 450 ul. |
| | | | |
| | ====Cutting the Oligo Pool==== | | ====Cutting the Oligo Pool==== |
| - | We decided to try cutting a pool of our 18 oligos. We used our new recipe, adding 0.78 ul of each oligo so that the DNA takes up a total of 16 ul. We followed the [[#New restriction enzyme protocol| new protocol]] for one reaction, and the other digestion was left to run overnight. We will compare the results tomorrow. | + | We decided to try cutting a pool of our 18 oligos. We used our new recipe, adding 0.78 ul of each oligo so that the DNA takes up a total of 16 ul. We followed the new protocol for one reaction, and the other digestion was left to run overnight. We will compare the results tomorrow. |
| | | | |
| | ===Team TolC=== | | ===Team TolC=== |
| Line 261: |
Line 261: |
| | **'''NOTE. It's possible that the ECNR2delTolC strain was used, but that still doesn't explain the 800bp band using the colonies from the zeocin plate used for selecting the zeocin cassette insertions that led to the WN strain (Noah's PCR 8/9/2011). It's strange given that the colonies off that plate give different results for the TolC region flanked by the TolC_seq primers, given that the zeocin PCR shows that all colonies have the zeocin insert (primers are Zeocin_R and rpoZ_R)''' | | **'''NOTE. It's possible that the ECNR2delTolC strain was used, but that still doesn't explain the 800bp band using the colonies from the zeocin plate used for selecting the zeocin cassette insertions that led to the WN strain (Noah's PCR 8/9/2011). It's strange given that the colonies off that plate give different results for the TolC region flanked by the TolC_seq primers, given that the zeocin PCR shows that all colonies have the zeocin insert (primers are Zeocin_R and rpoZ_R)''' |
| | {| | | {| |
| - | |[[File:2011.8.TolCKanpresence(labeled).png|thumb|blah]] | + | |[[File:HARV2011.8.TolCKanpresence(labeled).png|thumb|blah]] |
| - | |} | + | |}</div> |
| - | | + | <div id='809' style="display:none"> |
| | ==August 9th== | | ==August 9th== |
| | ===Team Wolfe=== | | ===Team Wolfe=== |
| Line 284: |
Line 284: |
| | ====5-FOA concentration gradient results==== | | ====5-FOA concentration gradient results==== |
| | *Since 5-FOA is more than worth its weight in gold, we were hoping that lower concentrations than recommended in the protocol would work for inhibiting the growth of cells expressing pyrF/URA3. According to last night's plate reader experiment, concentrations as low as .1mM completely stop cell growth! | | *Since 5-FOA is more than worth its weight in gold, we were hoping that lower concentrations than recommended in the protocol would work for inhibiting the growth of cells expressing pyrF/URA3. According to last night's plate reader experiment, concentrations as low as .1mM completely stop cell growth! |
| - | [[File: 2011.08.09.pSR01(2) 5-FOA grad.png|thumb|none|NM+his, NM, and 5-FOA concentration gradient in selection strain+pSR01]] | + | [[File: HARV2011.08.09.pSR01(2) 5-FOA grad.png|thumb|none|NM+his, NM, and 5-FOA concentration gradient in selection strain+pSR01]] |
| | *Also, another nice growth curve showing pSR01's rescue of the selection strain in incomplete media: | | *Also, another nice growth curve showing pSR01's rescue of the selection strain in incomplete media: |
| - | [[File: 2011.08.09.NM.png|thumb|none|selection strain and pSR01 in NM media]] | + | [[File: HARV2011.08.09.NM.png|thumb|none|selection strain and pSR01 in NM media]] |
| | | | |
| | ===Team Wolfe-ZF Collaboration=== | | ===Team Wolfe-ZF Collaboration=== |
| Line 298: |
Line 298: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.9_midiprep.jpg|thumb|Nanodrop data for midiprep.]] | + | |[[File:HARV2011.8.9_midiprep.jpg|thumb|Nanodrop data for midiprep.]] |
| | |} | | |} |
| | | | |
| Line 306: |
Line 306: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.9_plasmids_annotated.png|thumb|Miniprep vs. midiprep plasmid with equal loading volumes.]] | + | |[[File:HARV2011.8.9_plasmids_annotated.png|thumb|Miniprep vs. midiprep plasmid with equal loading volumes.]] |
| | |} | | |} |
| | | | |
| Line 314: |
Line 314: |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.9_oligo_5_6_sequencing_junction_pcr_annotated.png|thumb|Bands expected at around 1.5 kb instead of the usual 1.4]] | + | |[[File:HARV2011.8.9_oligo_5_6_sequencing_junction_pcr_annotated.png|thumb|Bands expected at around 1.5 kb instead of the usual 1.4]] |
| | |} | | |} |
| | | | |
| | ====Making more practice plate oligos==== | | ====Making more practice plate oligos==== |
| - | We were running low on oligos 1 through 6 from our practice plate, so we did a PCR to make more, using [[Lab_Notebook:_July#PCR_with_Practice_Plate| the same protocol as before]]. We then PCR purified the product. | + | We were running low on oligos 1 through 6 from our practice plate, so we did a PCR to make more, using the same protocol as before. We then PCR purified the product. |
| | | | |
| | {| | | {| |
| - | |[[File:2011.8.9_pcr_pure.jpg|thumb|Results of our PCR purification]] | + | |[[File:HARV2011.8.9_pcr_pure.jpg|thumb|Results of our PCR purification]] |
| | |} | | |} |
| | | | |
| Line 332: |
Line 332: |
| | Regardless, we performed a PCR purification on the product, and got a concentration of ~17 ng/ul. | | Regardless, we performed a PCR purification on the product, and got a concentration of ~17 ng/ul. |
| | | | |
| - | Then, we performed a ligation with the Oligo Pool and the CB Bottom backbone using the [[Lab_Notebook:_August#Practice_plate_ligation|typical protocol]]. We performed a chemical transformation with the ligation product, plating at two densities using 50 ul of transformed bacteria and 450 ul. We shall see tomorrow whether any colonies grew. | + | Then, we performed a ligation with the Oligo Pool and the CB Bottom backbone using the typical protocol. We performed a chemical transformation with the ligation product, plating at two densities using 50 ul of transformed bacteria and 450 ul. We shall see tomorrow whether any colonies grew. |
| | | | |
| | ====Yogurt Plate==== | | ====Yogurt Plate==== |
| Line 356: |
Line 356: |
| | *need to discard the WS and WN glycerol stocks into a 'bad glycerol stocks' box | | *need to discard the WS and WN glycerol stocks into a 'bad glycerol stocks' box |
| | *need to move the rpoZ MAGE oligos into the bad primers box | | *need to move the rpoZ MAGE oligos into the bad primers box |
| - | [[File: KNKSplatereadergood.png|thumb|none|Plate Reader of KN and KS at 2% SDS and varying IPTG conc. 8/9/11]] | + | [[File: HARVKNKSplatereadergood.png|thumb|none|Plate Reader of KN and KS at 2% SDS and varying IPTG conc. 8/9/11]] |
| | | | |
| | ====OZ052 and OZ123 MAGE into ECNR2+Kan-ZFB-wp+zeocin without plasmid==== | | ====OZ052 and OZ123 MAGE into ECNR2+Kan-ZFB-wp+zeocin without plasmid==== |
August 6th
Team ZF
New miniprep protocol
Today, we tried a new miniprep protocol in which the DNA is precipitated out of solution with EtOH, and no spin columns are used. This is because we suspect that our low yields may be due in some part to DNA being washed out of the column prematurely, or DNA not being washed completely out of the column during the elution step. In getting rid of the column, there is no step during which we may lose DNA.
We are extremely happy to report that after weeks of getting dismal miniprep yields, we have found a new protocol that gives us insanely high yields (compared to before)!!! The protocol is the following:
- Centrifuge 1.5 ml of saturated cells at maximum speed for 1 minute, discard supernatant.
- Resuspend cells in 200 ul of Buffer P1.
- Add 200 ul of Buffer P2 to lyse the cells. Mix by inverting several times, and let sit for 3 minutes.
- Add 200 ul of Buffer N3 to precipitate SDS, mix by inverting several times.
- Centrifuge at 4 degrees for 15 minutes.
- Transfer supernatant to new tube, add 1 ml of 100% EtOH to precipitate DNA.
- Centrifuge for 5 minutes at RT. At this point a DNA pellet should be visible at the bottom of the tube.
- Discard EtOH, wash DNA pellet with 1.5 ml of 70% EtOH (this gets rid of salts).
- Centrifuge for 5 minutes at RT.
- Discard EtOH, let tubes dry for approximately 6-8 minutes by inverting them on a piece of Kimwipe tissue.
- Resuspend DNA pellet in 20 ul of heated EB buffer.
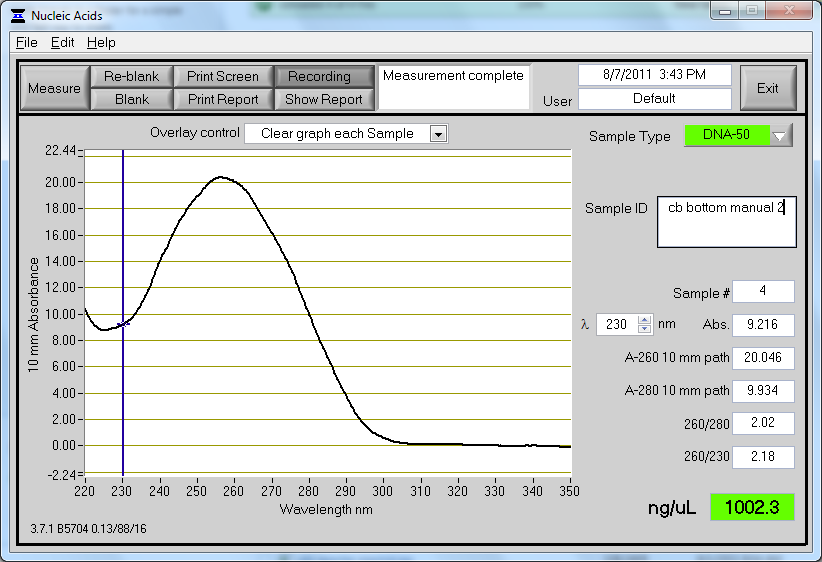
Our resulting yield was overall 463.4 ng/ul!! The purity also looked extremely good, with a 260/280 value of 1.98. The nanodrop results can be viewed below:
 Over 20x times normal yield compared with the Qiagen miniprep kit. |
We will have to repeat this again tomorrow to see if we get the same yields with this procedure. Also, we should run some of these out on a gel to verify what it looks like.
New restriction enzyme protocol
The protocol used for the restriction enzyme digest will be as described yesterday:
- 16 ul DNA (any concentration)
- 2 ul 10x buffer 4
- 0.5 ul 100x BSA
- 1.5 ul enzyme (30 units)
This gives a 20 ul total reaction volume. The reaction is incubated at 37* for 2 hours, denatured at 65* for 20 minutes, and then left at 4* for storage overnight.
A restriction digest using this protocol was performed on practice plate oligos 5 and 6 (reactions for oligos 3 and 4 were no good due to a pipetting mishap).
Sequencing results for the GFP reporter plasmid
Due to Seqman crashing and the Harvard VPN not being active, we will have to wait until Monday for these results.
Team Wolfe
MAGE round 3 allele-specific PRC:
- did 4 types of PCR: HisB KO (HisB VE KO check_R and His_F), HisB WT (HisB VE WT check_R and His_F), PyrF KO (PyrF KO check_F and PyrF_internal_R), PyrF WT (PyrF WT check_F and PyrF_internal_R)
- 24 colonies: A10, A11, C2, C3, C6, C8, C9, D1, D5-12, E3, E7, F2-4, G2, G3, G9
- 10uL total reaction volume
- same protocol as earlier reaction (64C anneal, 1min elongation, 25 cycles)
- HisB bands at about 900bp; PyrF bands at about 400bp.
- Results: none of the colonies have both genes knocked out but some do look like they may have the HisB deletion. We may try again using MAGE round 5 colonies.
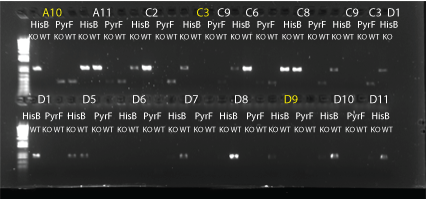
HisB and PyrF allele specific PCRs part 1. Yellow-labeled colonies may have a knock out. 8/6/11

Separate E gel of the pyrF PCRs cut off in gel 1. 8/6/11

HisB and PyrF allele specific PCRs part 2 8/6/11
pSB4K5 digestion:
- Since our minipreps have been giving such low yields, we tried an [http://openwetware.org/wiki/Knight:Miniprep_low_copy_plasmids altered protocol]. The result, however was the same: 6mL of culture produced 19ng/uL (in 30uL)
- PCR purified Spe1 Xba1 ZFB-wp-his3-ura3 digestion (Minelute): 53.8ng/uL 260/280=1.82. This is interesting, since it appears that half our DNA has been lost.
- pSB4K5 Spe1 and Xba1 digestion: 72uL total
- about 1mg DNA=60uL
- 2uL each enzyme
- 7.2uL buffer 4
- .72uL BSA
- 3 hour incubation at 37C
- gel extraction: the gel was empty. No clue why. Obviously this is a setback--we will have to redo the miniprep and digestion before we can form the whole plasmid.
August 7th
Team ZF
Miniprep repeat with new protocol
Another miniprep was repeated on 3 ml of CB bottom plasmid culture using the protocol from yesterday, with a yield of 1 ug/ul. This seemed very high considering our previous results, and we are suspicious that there may be some genomic DNA contamination. Contamination may occur either from leaving cultures on the shaker for too long (with cells lysing and releasing their genomic DNA) or from shaking too hard during the lysis process (shearing chromosomal DNA and allowing it to escape the cell). The lysis process was performed by mixing through tube inversion, so this is likely not the cause. The tubes that were miniprepped yesterday were left on the shaker for approximately 22 hours, while the tubes from today were on the shaker for 17 hours. Strangely, although the tubes were on the shaker for a shorter amount of time, there was still higher yield.
The concentration and absorbance curve data can be seen below:
 Hopefully this is actually real, it seems almost too good to be true. |
As a note, the tubes were unusually dark blue this round after addition of the lysis buffer (buffer P2).
The uncut plasmid was also run out on a gel alongside the cut oligos (loaded 0.5 ul). This should theoretically have been around 500 ng of DNA, but for some reason it only shows up very faintly. This is not good news.
Results for new restriction enzyme protocol
The resulting reaction from the oligo cut with Bsa-HF yesterday was run out on a gel with the following results:
There appears to be a band of the correct size. However, there is still incomplete cutting as indicated by the second band at around 100 bp, but there is no longer a band at the uncut position, indicating that all of the oligos underwent a single or double cut.
The plasmid DNA from the miniprep today was also put through a restriction digest using the same 2 hour protocol as the oligo digest. The result was then PCR purified, but the yield was extremely low compared to the input. Using 16 ul of DNA, there were 16 ug of plasmid that went into the restriction digest. After PCR purification and elution in 30 ul of EB, the yield was only 28.5 ng/ul, meaning only 0.855 ug of DNA in the purified product. This translate to over 95% of the DNA somehow being lost in the PCR purification. We are at a complete loss as to why this has happened...
The cut plasmid DNA was run alongside uncut plasmid in a gel, with the results below:
Although the bands are faint, the cut band is somewhere around the correct size (around 2.8 kb). Although the band appears to be anywhere between 3 kb and 4 kb in the gel, the ladder ran strangely for some reason. However, there is still a fairly well-defined band instead of a smear in the cut lane. This in itself is a huge relief - if there were significant genomic DNA contamination, we might expect to get a smear from many differently sized cuts. Furthermore, the uncut band looks standard for a plasmid, similar to the picture on the website here:
http://faculty.plattsburgh.edu/donald.slish/Electrophoresis.html. There are three bands in our gel as they appear on the website. However, our linearized plasmid is actually fairly close to the bottom band for the uncut plasmid, which is presumably the supercoiled form.
August 8th
Team Wolfe
MAGE sequencing
- Colonies
- A10, C3, and D9 from round 3 MAGE
- A1-D8 round 5 MAGE (colonies picked today).
- Primers for PCR
- His_F, His_R
- PyrF_F, PyrF_R
- Primers for sequencing
- 64 degrees annealing and 90 seconds elongation
- Gel shows PCR was successful
5-FOA Concentration Gradient
- Protocol recommended 2.5mM conc. FOA but we tested lower concentrations because FOA = $$$: ($FOA>$Gold)
- 61,68...89 NM+His
- 62,69...90 NM
- 63,70...91 .1mM 5-FOA
- 64,71...92 .25mM 5-FOA
- 65,72...93 .5mM 5-FOA
- 66,73...94 1mM 5-FOA
- 67,74...95 2.5mM 5-FOA
- 61-67 = selection strain colony 1
- 68-74 = selection strain colony 2
- 75-81 = pSR01 colony 1
- 82-88 = pSR01 colony 2
- 89-95 = empty
pSB4K5
- Since minipreps are not working we will try a midiprep
- Innoculated 100mL LB+Kan with a pSB4K5 3:11P colony
Streaked Plates
- Selection strain
- Selection strain + pSR01 plates because they were getting old
Team ZF
GFP Sequencing Results
We were able to access the results of our GFP sequencing.
|
| GFP Forward
| Notes
|
| | 1 | 1 deletion at end
|
| ✓ | 2 | perfect
|
| | 3 | Insert of G
|
| | 4 | perfect
|
|
| GFP Reverse
| Notes
|
| | 1 | 4 snps
|
| ✓ | 2 | 1 deletion at end
|
| | 3 | 0 snps
|
| | 4 | snps
|
However our sequencing failed to cover the the Shine-Dalgarno region. We decided to test our strains by growing them on a 96 well plate. Each well had 150 ul LB+spec. Colonies designated 1-4 are 5 ul of the ones we sent out for sequencing. Those designated 5-8 were 2 ul colonies which were picked from our reporter plate then and diluted in 10 ul LB+spec. The plate had the following configuration:
- Rows A and B, 1 through 8: New colonies picked from the plate of our reporter strain.
- Row E, 1 through 8: Colonies 1-8 in 0 ul IPTG
- Row F, 1 through 8: Colonies 1-8 in 2 ul IPTG
- Row G, 1 through 8: Colonies 1-8 in 10 ul IPTG
- Row H, 1 through 8: Colonies 1-8 in 100 ul IPTG
After shining blue light on the plate, nothing glowed. We realized that this was because we did not have a spacer between the Shine-Dalgarno sequence and the start codon, meaning the ribosome would not be able to reach both sites. After doing some reading, we realized that a spacer of 6-10 bps is ideal. We will design this tomorrow.
Re-do minipreps for control backbones with new protocol
We used the new protocol to do a miniprep on the control backbones.
 We got (unusually?) high yields using the new technique. |
Our yields were extremely high with the new technique--it was difficult to believe. We were a bit wary of the nanodrop readings so we decided to run the product on a gel and compare the brightness of the band to the ladder in order to determine the actual concentration of DNA present in the miniprep product.
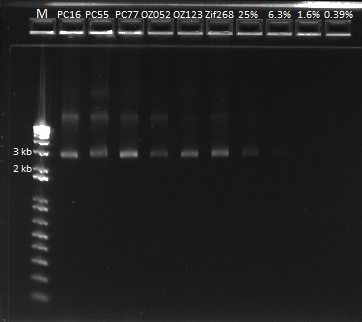 We ran a gel of our new miniprep product. |
The first six lanes are our miniprep product. The next four contain serial dilutions of the Zif 268 plasmid. We determined that it looks like our miniprep product actually contains ~80 ng/ul of DNA, after comparing it with the 1650 bp band.
Ligation and transformation of Oligo 5 and 6 and CB Bottom
We decided to try the ligation of our oligos and the backbone, using our previous protocol. We then transformed the product using ChemComp cells, plating 50 and 450 ul.
Cutting the Oligo Pool
We decided to try cutting a pool of our 18 oligos. We used our new recipe, adding 0.78 ul of each oligo so that the DNA takes up a total of 16 ul. We followed the new protocol for one reaction, and the other digestion was left to run overnight. We will compare the results tomorrow.
Team TolC
Plate Reader
- Set up and began running 160 well plate reader
- Plate was set up as follows:
PCR on TolC
- Have failed twice before in trying to PCR out the TolC sequence from the WS and WN strains, which are ECNR2 without the Kan-ZFB-wp insert, worried that TolC may have somehow been deleted or removed which seems very unlikely
- Ran third PCR and gel and failed again
- NOTE. It's possible that the ECNR2delTolC strain was used, but that still doesn't explain the 800bp band using the colonies from the zeocin plate used for selecting the zeocin cassette insertions that led to the WN strain (Noah's PCR 8/9/2011). It's strange given that the colonies off that plate give different results for the TolC region flanked by the TolC_seq primers, given that the zeocin PCR shows that all colonies have the zeocin insert (primers are Zeocin_R and rpoZ_R)
August 9th
Team Wolfe
Sequencing check for HisB and PyrF deletions
- One colony definitely has both deletions! It is now being grown for lambda red and glycerol stock
Zeocin Lambda Red
- Made 10 zeocin plates (zeocin concentration=50µg/mL)
- Grew up cells from D2 MAGE#5 to midlog and used 1mL for lambda red
- 2µL zeocin cassette (about 300ng)
- recovered for 2 hours before plating
pSB4K5 Digestion
- Enzymes
- DNA = pSB4K5 plasmid (1µg, 17.2µL)
- 4µL buffer 4
- .4µL BSA (100x)
- 14.4µL H2O
- incubated for 2hrs at 37C
5-FOA concentration gradient results
- Since 5-FOA is more than worth its weight in gold, we were hoping that lower concentrations than recommended in the protocol would work for inhibiting the growth of cells expressing pyrF/URA3. According to last night's plate reader experiment, concentrations as low as .1mM completely stop cell growth!

NM+his, NM, and 5-FOA concentration gradient in selection strain+pSR01
- Also, another nice growth curve showing pSR01's rescue of the selection strain in incomplete media:

selection strain and pSR01 in NM media
Team Wolfe-ZF Collaboration
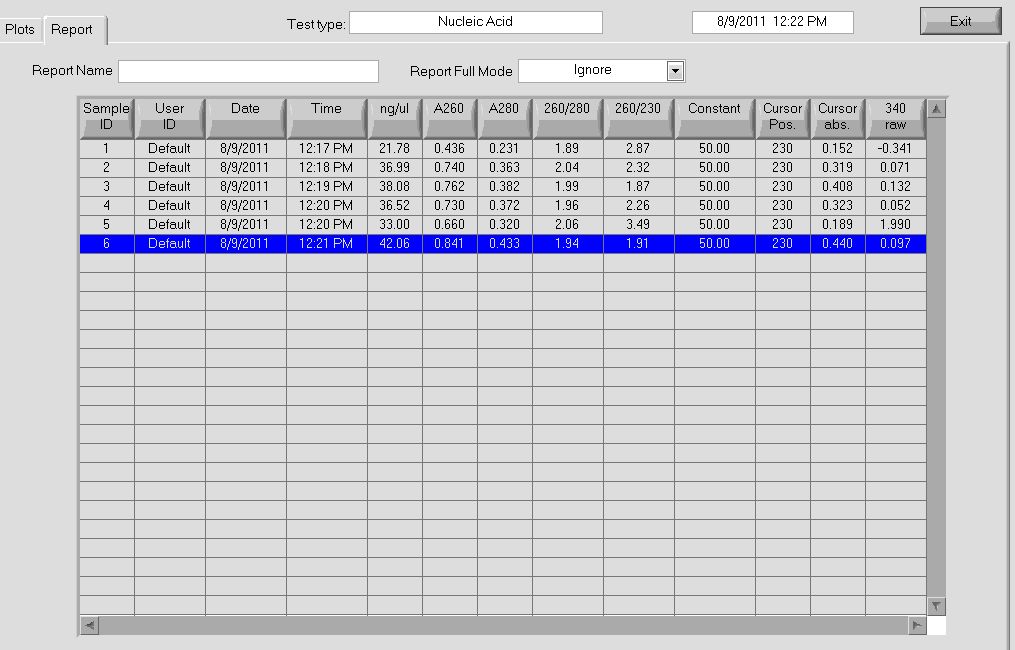
Midiprep
- Note: Instead of second lysate clearing spin, supernatant was transferred to falcon tube and poured into column
- Yields
- pSB4K5 = 58ng/µL, 260/280=1.84
- Zif268 = 168.52 ng/ul, 260/280 = 1.84
More data on the concentrations can be seen below:
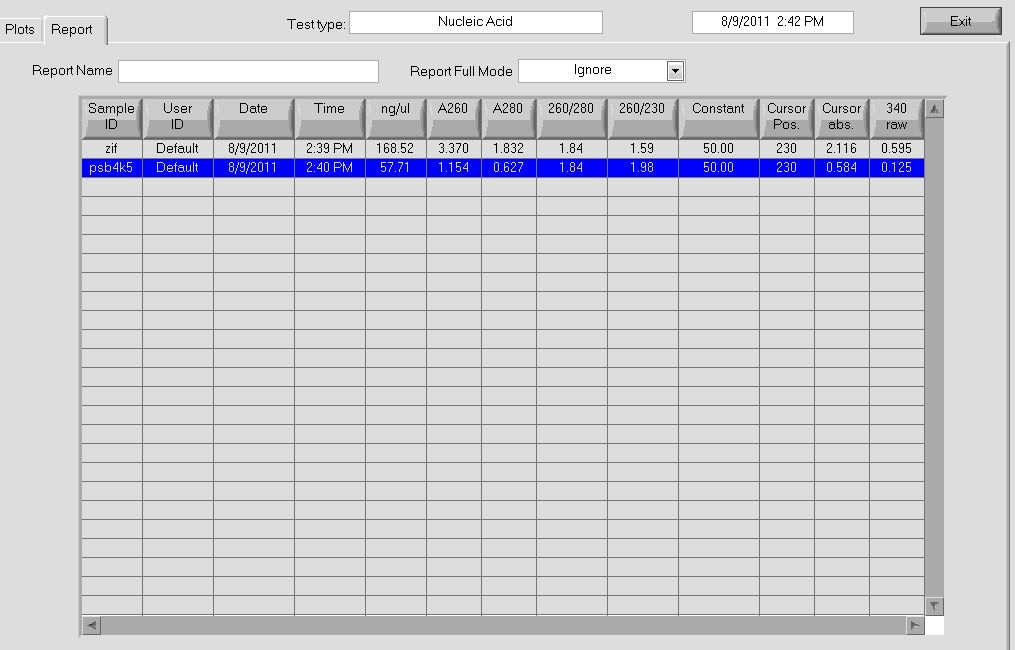 Nanodrop data for midiprep. |
The bands for the midiprep look similar to those from the miniprep using the new ethanol precipitation protocol, except they are much brighter. In the following gel image, there were 8 ul of plasmid loaded for both the miniprep and midiprep plasmids. It appears that there is much, much less miniprep plasmid than midiprep. Considering that the midiprep plasmid is at approximately 168.52 ng/ul, the miniprep plasmid is probably around our old yield. This means that the Nanodrop reader LIED TO US!! :'( :'( :'(
Here is the gel:
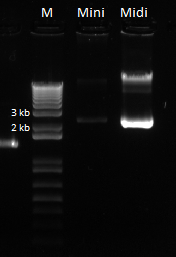 Miniprep vs. midiprep plasmid with equal loading volumes. |
Team ZF
Checking our 5 and 6 Ligation Plate
Colonies grew on all our plates! We ran our cross-junction PCR on 8 colonies each from the 450 ul plates and sent it out for sequencing.
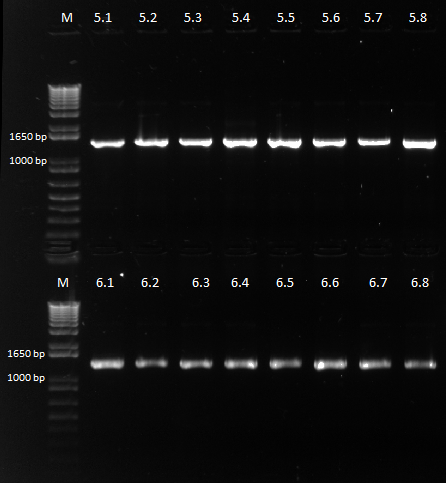 Bands expected at around 1.5 kb instead of the usual 1.4 |
Making more practice plate oligos
We were running low on oligos 1 through 6 from our practice plate, so we did a PCR to make more, using the same protocol as before. We then PCR purified the product.
 Results of our PCR purification |
Design a new primer for GFP
We designed a new primer for GFP, with a 7 bp spacer between the Shine-Dalgarno sequence and the start codon for GFP. According to what we discovered online, it seems that the spacing between the ribosome binding site and the ATG start codon is very important for transcription efficiency. We had previously thought that there were 8 bp from the beginning of the RBS to the ATG codon, but instead it is really 5-9 bp in between the end of the RBS and the ATG codon that is correct. We thus added the spacer that comes directly from the Ambion website, which is AGGAGGTATATACATG (spacer bolded). More info can be found here: http://www.ambion.com/techlib/basics/translation/index.html.
Practice plate oligo pool
This morning, we took out our digestion reactions from the thermocycler, containing all 18 practice plate target oligos mixed together. The tube containing the overnight reaction was a bit crushed by the machine, and it looks like we lost a bit of volume.
Regardless, we performed a PCR purification on the product, and got a concentration of ~17 ng/ul.
Then, we performed a ligation with the Oligo Pool and the CB Bottom backbone using the typical protocol. We performed a chemical transformation with the ligation product, plating at two densities using 50 ul of transformed bacteria and 450 ul. We shall see tomorrow whether any colonies grew.
Yogurt Plate
Yesterday we plated 10 ul of [http://www.stonyfield.com/products/stonyfield/smoothies/10oz-smoothies/peach-0 Stonyfield Super Smoothie] from lunch, which contains [http://www.stonyfield.com/products/stonyfield/what-makes-our-products-so-special/six-live-cultures six varieties of active yogurt bacteria]. Today it grew colonies. It smelled like yogurt this morning. This evening it smelled like rotting yogurt. Yuck.
(UPDATE: According to Dan it smells like Swiss cheese. Now it smells slightly better).
Prepping for western blot tomorrow
We inoculated cultures (4 ml each) in preparation for western blotting tomorrow to verify that the zinc fingers are indeed being expressed by IPTG. Here is the setup:
- Kan - pze22g with GFP as a complete negative control (there should be no zinc fingers in there... hoping there's also no proteins with flag tags in there)
- Spec - Zif268 whole with 0 uM IPTG
- Spec - Zif268 whole with 2 uM IPTG
- Spec - Zif268 whole with 10 uM IPTG
- Spec - Zif268 whole with 100 uM IPTG
We expect to see zinc finger expression to varying degrees in the cultures with IPTG, depending on the level of IPTG. Hopefully we'll have enough protein and FLAG tag antibody to work with tomorrow.
Team TolC
Plate Reader
- There appears to be a significant difference between the KN and KS strands, but only at high SDS levels i.e. 2% (lag of about 5 hours). At lower levels, there appears to be no significant lag, but a difference in the slope of the log phase.
- Even at 0 IPTG, there is expression of the ZF plasmid, suggesting leakiness (constitutively 'ON' promoter for the ZFP)
- WS and WN strands show lack of growth in the presence of SDS at any concentration (except 0) and PCR shows odd results (no band to 800bp bands for the product using tolC_seq_F and tolC_seq_R) add images here
- need to discard the WS and WN glycerol stocks into a 'bad glycerol stocks' box
- need to move the rpoZ MAGE oligos into the bad primers box
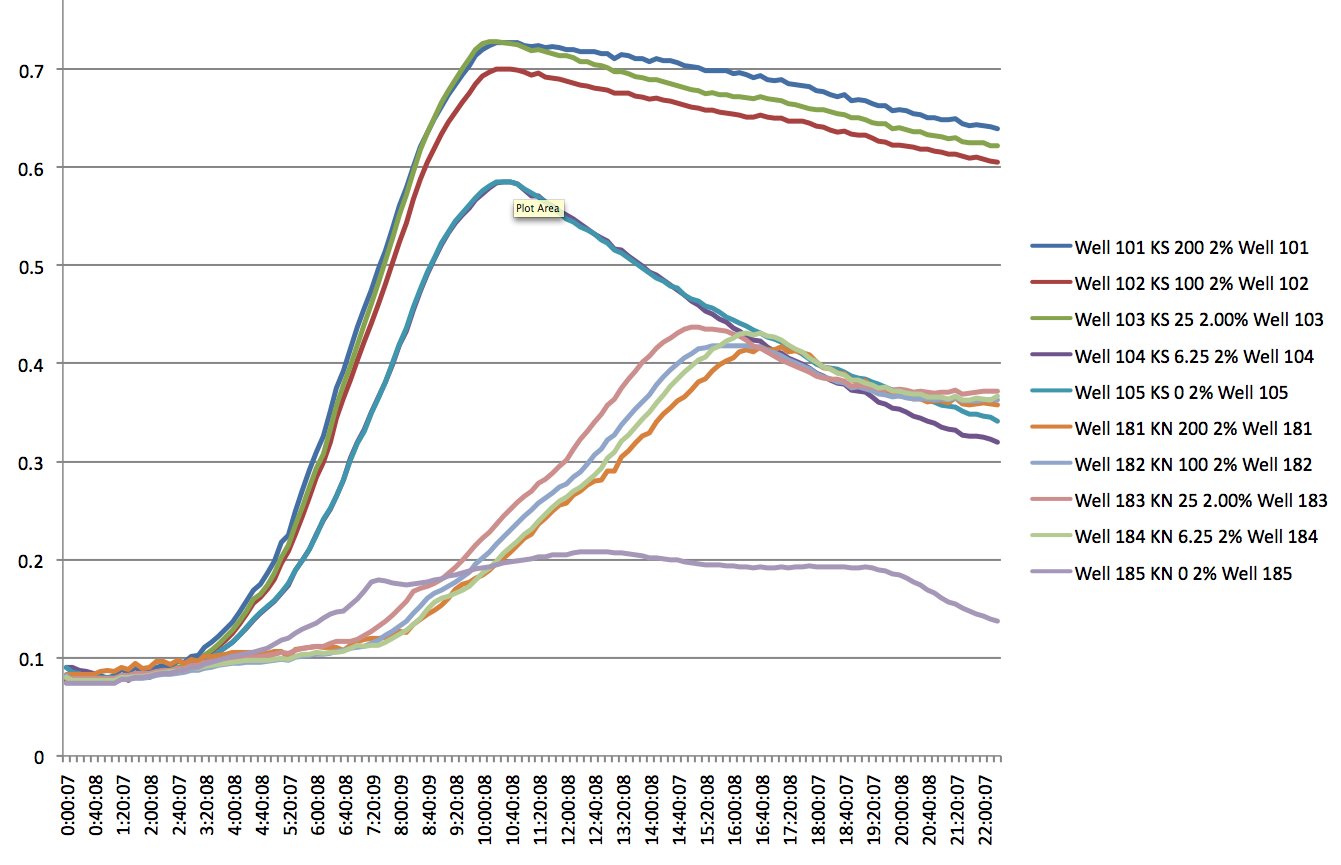
Plate Reader of KN and KS at 2% SDS and varying IPTG conc. 8/9/11
OZ052 and OZ123 MAGE into ECNR2+Kan-ZFB-wp+zeocin without plasmid
- Ran two rounds of MAGE for both OZ052 and OZ123 in the KN strain order to create the other two positive controls.
- time constants were 5.2s for the first round, and 5.6s for the second round. After the second round of MAGE, the cells appear to have been growing rather slowly. MAGE 2 was done at 2pm, and the cultures are barely cloudy at 6:28pm
- 3rd round at 10:10pm, time constants are 5.4s and 4.6s
Lambda Red Recombination of zeocin into new ECNR2 culture for rpoZ knockout
- put the colony in 2ml of LB+amp to grow at 3pm. It should be ready for lambda red by 7pm, hopefully?
 "
"

-0016.png)
-0018.png)
-0017.png)



-0019.png)


.png)
_5-FOA_grad.png)





