 "
"
Team:TU Munich/lab/notebook/part3
From 2011.igem.org
| (14 intermediate revisions not shown) | |||
| Line 1: | Line 1: | ||
<html> | <html> | ||
<script src="https://2011.igem.org/Team:TU_Munich/slimbox2.js?action=raw&ctype=text/js" type="text/javascript"></script> | <script src="https://2011.igem.org/Team:TU_Munich/slimbox2.js?action=raw&ctype=text/js" type="text/javascript"></script> | ||
| - | <link rel="stylesheet" href="https://2011.igem.org/Team:TU_Munich/slimbox2.css?action=raw&ctype=text/css" type="text/css" media="screen"> | + | <link rel="stylesheet" href="https://2011.igem.org/Team:TU_Munich/slimbox2.css?action=raw&ctype=text/css" type="text/css" media="screen"></link> |
<div class="ui-corner-all subcontent tabcontent"> | <div class="ui-corner-all subcontent tabcontent"> | ||
| - | <h1><span class="mw-headline" id="Light_sensor_systems_and_AND-Gate_cloningPart_III_Alex.2FSimon.2FBea.2FThorsten">Cloning Part III</span></h1> | + | |
| + | <h1><span class="mw-headline" id="Light_sensor_systems_and_AND-Gate_cloningPart_III_Alex.2FSimon.2FBea.2FThorsten">Cloning Part III + Results</span></h1> | ||
<p><b>People: Alex, Simon, Bea, Thorsten, Anna</b></p> | <p><b>People: Alex, Simon, Bea, Thorsten, Anna</b></p> | ||
| + | |||
<br> | <br> | ||
| Line 580: | Line 582: | ||
<div class="cloning"> | <div class="cloning"> | ||
<h4><span class="mw-headline" id="Glycogen.2FEtOH-precipitation_of_ligation_samples_from_10-09-11_and_transformation">Glycogen/EtOH-precipitation of ligation samples from 10-09-11 and transformation</span></h4> | <h4><span class="mw-headline" id="Glycogen.2FEtOH-precipitation_of_ligation_samples_from_10-09-11_and_transformation">Glycogen/EtOH-precipitation of ligation samples from 10-09-11 and transformation</span></h4> | ||
| - | <p>Put everything on ice before starting to work! Pre-cool centrifuge!</p><p>See protocol for precipitation/transformation from 07-09-11[ | + | <p>Put everything on ice before starting to work! Pre-cool centrifuge!</p><p>See protocol for precipitation/transformation from 07-09-11[See Methods]</p><p>Changes made:</p> |
<ul><li>all solutions were icecold</li><li>pellet was air-dryed for about 10 min</li><li>autoclaved water was used instead of nuclease-free, due to quick subsequent use in the transformation</li><li>resuspending of the bacterial pellet after transformation was difficult (tried pipetting and vortexing), because the wrong centrifuge has been used</li><li>1,5 ml-Eppis were used instead of 2 ml-Eppis for the shaking step at 37°C in SOC</li></ul> | <ul><li>all solutions were icecold</li><li>pellet was air-dryed for about 10 min</li><li>autoclaved water was used instead of nuclease-free, due to quick subsequent use in the transformation</li><li>resuspending of the bacterial pellet after transformation was difficult (tried pipetting and vortexing), because the wrong centrifuge has been used</li><li>1,5 ml-Eppis were used instead of 2 ml-Eppis for the shaking step at 37°C in SOC</li></ul> | ||
<p><b>Samples 1, 8 and 9</b>: comp. cells used were DH5a from <b>01-07-11</b></p><p><b>Samples 2 - 7</b>: comp. cells used were DH5a from <b>30-08-11</b></p><p>Antibiotic resistance: </p> | <p><b>Samples 1, 8 and 9</b>: comp. cells used were DH5a from <b>01-07-11</b></p><p><b>Samples 2 - 7</b>: comp. cells used were DH5a from <b>30-08-11</b></p><p>Antibiotic resistance: </p> | ||
| Line 634: | Line 636: | ||
<p><b>People: Simon, Anna, Alex, Thorsten</b></p> | <p><b>People: Simon, Anna, Alex, Thorsten</b></p> | ||
<h3><span class="mw-headline">Cloning</span></h3> | <h3><span class="mw-headline">Cloning</span></h3> | ||
| + | |||
<div class="cloning"> | <div class="cloning"> | ||
| + | |||
| + | <p>One colony from yesterday's transformation of Part I746907 in pSB1A2 was picked and inoculated into 5 ml LB Amp. Incubation o/n at 37 °C</p> | ||
| + | |||
<h4><span class="mw-headline" id="AND-Gate">AND-Gate</span></h4> | <h4><span class="mw-headline" id="AND-Gate">AND-Gate</span></h4> | ||
<p>Minipreps of picked cones on 12-09-11 were done according to Metabion's instructions. Concentrations:</p><p>2a: c = 68.5 ng/µl</p><p>2b: c = 68.0 ng/µl</p><p>3a: c = 118 ng/µl</p><p>3b: c = 65.0 ng/µl</p><p>5a: c = 122 ng/µl</p><p>5b: c = 71.5 ng/µl</p><p>6a: c = 122 ng/µl</p><p>6b: c = 119 ng/µl</p><p>6c: c = 51.0 ng/µl</p><p>After restriction digest using E and P, samples were separated on a 0.8 % gel:</p><p><a href="/wiki/images/5/51/110913_analytischer_verdau_and-gate_Thorsten.jpg" class="image" rel="lightbox"><img alt="110913 analytischer verdau and-gate Thorsten" src="/wiki/images/5/51/110913_analytischer_verdau_and-gate_Thorsten.jpg" width="400" /></a></p> | <p>Minipreps of picked cones on 12-09-11 were done according to Metabion's instructions. Concentrations:</p><p>2a: c = 68.5 ng/µl</p><p>2b: c = 68.0 ng/µl</p><p>3a: c = 118 ng/µl</p><p>3b: c = 65.0 ng/µl</p><p>5a: c = 122 ng/µl</p><p>5b: c = 71.5 ng/µl</p><p>6a: c = 122 ng/µl</p><p>6b: c = 119 ng/µl</p><p>6c: c = 51.0 ng/µl</p><p>After restriction digest using E and P, samples were separated on a 0.8 % gel:</p><p><a href="/wiki/images/5/51/110913_analytischer_verdau_and-gate_Thorsten.jpg" class="image" rel="lightbox"><img alt="110913 analytischer verdau and-gate Thorsten" src="/wiki/images/5/51/110913_analytischer_verdau_and-gate_Thorsten.jpg" width="400" /></a></p> | ||
| Line 720: | Line 726: | ||
<div class="cloning"> | <div class="cloning"> | ||
<h4> <span class="mw-headline" id="Miniprep">Miniprep</span></h4> | <h4> <span class="mw-headline" id="Miniprep">Miniprep</span></h4> | ||
| - | <p>Part I746907 (pSB1A2) was purified from yesterday's DH5alpha-pellet using the miniprep-kit according to protocol.</p> | + | <p>Part I746907 (pSB1A2) was purified from yesterday's DH5alpha-pellet using the miniprep-kit according to protocol. The concentration was determined to be 63 ng/µl using Nanodrop.</p> |
<h4> <span class="mw-headline" id="Glycogen.2FEthanol_precipitation_and_Transformation_2">Glycogen/Ethanol precipitation and Transformation</span></h4> | <h4> <span class="mw-headline" id="Glycogen.2FEthanol_precipitation_and_Transformation_2">Glycogen/Ethanol precipitation and Transformation</span></h4> | ||
<p>Yesterday's ligations were purified using glycogen/ethanol precipitation. The yielded 10 µl DNA-Solution were used completely for Transformation in DH5alpha. After electroporation, the cells were incubated at 37 °C for 1h in SOC-media. After this, they were plated onto LB-Agar plates with the appropriate antibiotics.</p> | <p>Yesterday's ligations were purified using glycogen/ethanol precipitation. The yielded 10 µl DNA-Solution were used completely for Transformation in DH5alpha. After electroporation, the cells were incubated at 37 °C for 1h in SOC-media. After this, they were plated onto LB-Agar plates with the appropriate antibiotics.</p> | ||
| Line 727: | Line 733: | ||
Our synthetic construct (in pMA-RQ) was digested for preparation using E and S, while the miniprepped part I746907 was digested using E and S to check if the prepped plasmids are correct (a mistake was made during preparation of the digest, so a second digest was made at the same time. Both digests were loaded onto the analytical gel):</p><p><br /> | Our synthetic construct (in pMA-RQ) was digested for preparation using E and S, while the miniprepped part I746907 was digested using E and S to check if the prepped plasmids are correct (a mistake was made during preparation of the digest, so a second digest was made at the same time. Both digests were loaded onto the analytical gel):</p><p><br /> | ||
<a href="/wiki/images/8/8c/Digestions150911.jpg" class="image" rel="lightbox"><img alt="Digestions150911" src="/wiki/images/8/8c/Digestions150911.jpg" width="400" /></a></p><p>The product of the preparative digest was loaded onto a preparative 1% agarose gel. After electrophoresis, the desired band was cut out on the UV-transilluminator (365 nm) before the gel photo was recorded:</p><p><br /> | <a href="/wiki/images/8/8c/Digestions150911.jpg" class="image" rel="lightbox"><img alt="Digestions150911" src="/wiki/images/8/8c/Digestions150911.jpg" width="400" /></a></p><p>The product of the preparative digest was loaded onto a preparative 1% agarose gel. After electrophoresis, the desired band was cut out on the UV-transilluminator (365 nm) before the gel photo was recorded:</p><p><br /> | ||
| - | The product of the control digest was loaded onto an analytic 0.8 | + | The product of the preparative digest was loaded onto a preparative 1 % agarose gel. After electrophoresis, the desired band was cut out on the UV-transilluminator (365 nm) before the gel photo was recorded: :</p> |
| + | <a href="https://static.igem.org/mediawiki/2011/e/ed/15092011_preparative_gel_synthetic_construct.jpg" class="image" rel="lightbox"><img alt="Digestions150911" src="https://static.igem.org/mediawiki/2011/e/ed/15092011_preparative_gel_synthetic_construct.jpg" width="400" /></a> | ||
| + | <p>The gel looks as expected. The cut out bands around 400 bp are the synthetic construct while the vector can be seen at 2.4 kb.</p> | ||
| + | <p>The product of the control digest was loaded onto an analytic 0.8 % agarose gel. The following gel photo was recorded:</p> | ||
| + | <a href="https://static.igem.org/mediawiki/2011/4/4a/15092011_analytical_gel_I746907.jpg" class="image" rel="lightbox"><img alt="Digestions150911" src="https://static.igem.org/mediawiki/2011/4/4a/15092011_analytical_gel_I746907.jpg" width="400" /></a> | ||
| + | |||
| + | <p>Lane 1 (correct digest): Part BBa_I746907 is 924 bp long, the gel shows the corresponding bands. The vector pSB1A2 is 2079 bp long and can also be seen at the expected spot.</p> | ||
| + | |||
| + | <p>Lane 2 (wrong digest): The bands show that the digest doesn't work properly in 10x NEB4 buffer. The band around 3 kb is uncut or singly cut vector with insert.</p> | ||
</div> | </div> | ||
<h3> <span class="mw-headline">Testing</span></h3> | <h3> <span class="mw-headline">Testing</span></h3> | ||
<div class="testing"> | <div class="testing"> | ||
<h4> <span class="mw-headline" id="Miller_Assay_with_reporter_construct">Miller Assay with reporter construct</span></h4> | <h4> <span class="mw-headline" id="Miller_Assay_with_reporter_construct">Miller Assay with reporter construct</span></h4> | ||
| - | <p>two cultures in LB medium:</p> | + | <p>We incubated two cultures over night in LB medium with the appropriate antibiotics: |
| - | <ul><li> | + | </p> |
| - | </li><li> | + | <ul><li>BBa_K568003 (T7 promotor and β-galactosidase) in BL21(DE3); |
| + | </li><li>Negative control BBa_I732017 (rbs and β-galactosidase) in DH5 alpha. | ||
</li></ul> | </li></ul> | ||
| - | <p> | + | <p>The cultures were diluted to a OD_600 of about 0.7 and incubated for another 30 min. Then, the cultures were induced with IPTG at different concentrations: |
| - | + | </p> | |
| - | + | <ul><li>BBa_K568003: 0 mM, 0.01 mM, 0.1 mM, 1.0 mM and 1.5 mM; | |
| + | </li><li>BBa_I732017: 0 mM and 1.0 mM. | ||
| + | </li></ul> | ||
| + | <p>Before induction and every 30 min, the Miller Units were measured as follows: | ||
| + | |||
| + | </p> | ||
| + | <ol><li>measurement of Abs(600nm) in plate reader (volume of bacterial suspension should be equal in all wells, ideally 170 µl) | ||
| + | </li><li>new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS + 40 μl of Chloroform (under fume hood) + 500 µl sample solution (e.g. 430 µl medium + 20 µl bacterial suspension) | ||
| + | </li><li>mix the solution by vortexing for 10 s (all samples with equal vortexing time) | ||
| + | </li><li>let chloroform settle down (this takes about 5 min, if tube still contains blurred solution, centrifuge for 1 min, RT, 4000 rcf) | ||
| + | </li><li>transfer 100 μl of supernatant to 96 well plate (for photometer) | ||
| + | </li><li>initiation of assay with 20 μl of ONPG (4mg/ml), mix well, NOTE START TIME | ||
| + | </li><li>incubation at 37 °C | ||
| + | </li><li>stop reaction with 50 μl of 1 M Na2CO3, mix well, NOTE STOP TIME | ||
| + | </li><li>measurement of Abs (420nm) and Abs (550nm) | ||
| + | </li></ol> | ||
| + | <p>The Miller Units were calculated according to the following equation: | ||
| + | </p><p>Miller Units = 1000 * (ABS420 - (1.75 * ABS550)) / (time [min] * volume [ml] * ABS600) | ||
| + | </p><p>The reaction time was 5 min and the culture volume used was 10 µl. | ||
| + | </p><p><br /> | ||
| + | </p> | ||
| + | <h3> <span class="mw-headline">Other Work</span></h3> | ||
| + | <div class="otherwork"> | ||
</div> | </div> | ||
<h3> <span class="mw-headline">Other Work</span></h3> | <h3> <span class="mw-headline">Other Work</span></h3> | ||
<div class="otherwork"> | <div class="otherwork"> | ||
| - | <p>Testing the spectrometer (at the wavelengths 420 and 600 nm, because they are used in the Miller Assay: over which range are the OD values linear?)</p><p>samples:</p> | + | <p>Testing the spectrometer (at the wavelengths 420 and 600 nm, because they are used in the Miller Assay: over which range are the OD values linear?)</p> |
| - | <ul><li>for 600 nm measurements: 170 µl of a bacterial culture in LB medium, blank with fresh LB medium, dilutions in LB medium (minor problem: <b>cells could still grow during the experiment</b>, especially as the plate reader heated the plate) | + | <p>samples:</p> |
| - | </li><li>for 420 nm measurements: 130 µl of one sample of Miller Assay from 13-09-2011 (had already been developed/stopped and stored o/n at RT), blank with ddH2O, dilutions in ddH2O | + | <ul><li>for 600 nm measurements: 170 µl of a bacterial culture in LB medium, blank with fresh LB medium, dilutions in LB medium (minor problem: <b>cells could still grow during the experiment</b>, especially as the plate reader heated the plate)</li> |
| + | <li>for 420 nm measurements: 130 µl of one sample of Miller Assay from 13-09-2011 (had already been developed/stopped and stored o/n at RT), blank with ddH2O, dilutions in ddH2O | ||
</li></ul> | </li></ul> | ||
| + | <p><br /></p> | ||
</div> | </div> | ||
<h3> <span class="mw-headline">Results</span></h3> | <h3> <span class="mw-headline">Results</span></h3> | ||
<div class="results"> | <div class="results"> | ||
| + | <h4><span class="mw-headline" id="Miller_Assay_Results">Miller Assay Results</span></h4> | ||
| + | <p>The Miller Assay yielded the following data set:</p> | ||
| + | <p><a href="/wiki/images/9/97/Data_Miller_Assay_15-09-2011.png" class="image" rel="lightbox"><img alt="Data Miller Assay 15-09-2011.png" src="/wiki/images/9/97/Data_Miller_Assay_15-09-2011.png" width="400" /></a></p> | ||
| + | <p><a href="/wiki/images/6/65/TUM_2011_Miller_Assay_15.9.2011_of_BBa_K568003_Labbook_Version.png" class="image" rel="lightbox"><img alt="TUM 2011 Miller Assay 15.9.2011 of BBa K568003 Labbook Version.png" src="/wiki/images/6/65/TUM_2011_Miller_Assay_15.9.2011_of_BBa_K568003_Labbook_Version.png" width="400" /></a></p> | ||
| + | <p>The data shows that the cultures induced by isopropyl-β-D-1-thiogalactopyranoside (IPTG) yield higher Miller Units than the uninduced control after 60 min. Therefore, a higher amount of β-galactosidase was produced. This shows that the part is working as expected. Furthermore a slight quantitative dependence of the Miller Units on the concentration of IPTG can be seen. After 120 min the Miller Units of the culture induced with 1.0 mM IPTG go down again, which might be evidence for increasing of proteolysis caused by death of bacteria due to a too high concentration of IPTG. It can also be observed that the uninduced control yields higher Miller Units than the β-galactosidase negative, induced control. This can be explained by leaky biosynthesis of the genome-coded T7 polymerase. If the production of T7 polymerase is more tightly controlled, the level of expressed β-galactosidase should go down to the minimum of the negative, induced control.</p> | ||
| + | <h4><span class="mw-headline" id="Testing_of_plate_reader_photometer">Testing of plate reader photometer</span></h4> | ||
<p><b>linear over total range tested, results of yesterday are valid</b></p> | <p><b>linear over total range tested, results of yesterday are valid</b></p> | ||
| - | <ul><li>at 420 nm: OD = 0.15 - 3.10 | + | <ul><li>at 420 nm: OD = 0.15 - 3.10</li> |
| - | </li><li>at 600 nm: OD = 0.01 - 2.26 | + | <li>at 600 nm: OD = 0.01 - 2.26 |
</li></ul> | </li></ul> | ||
| - | <p><a href="/wiki/images/d/d8/Test_of_spectrometer_420.jpg" class="image" rel="lightbox"><img alt="Test of spectrometer 420" src="/wiki/images/d/d8/Test_of_spectrometer_420.jpg" width="400" | + | <p><a href="/wiki/images/d/d8/Test_of_spectrometer_420.jpg" class="image" rel="lightbox"><img alt="Test of spectrometer 420" src="/wiki/images/d/d8/Test_of_spectrometer_420.jpg" width="400" /></a></p> |
| + | <p><a href="/wiki/images/b/b1/Test_of_spectrometer.jpg" class="image" rel="lightbox"><img alt="Test of spectrometer" src="/wiki/images/b/b1/Test_of_spectrometer.jpg" width="400" /></a></p> | ||
</div> | </div> | ||
| - | <h2> <span class="mw-headline" id="16-09-2011">16-09-2011</span></h2> | + | <h2><span class="mw-headline" id="16-09-2011">16-09-2011</span></h2> |
| - | <p><b>People: Simon, Anna</b></p><p><br /></p> | + | <p><b>People: Simon, Anna</b></p> |
| + | <h3><span class="mw-headline" id="Results_of_15-09-2011">Results of 15-09-2011</span></h3> | ||
| + | <p>The plates of the transformation (products of the ligation from 14-09-2011) showed the following amount of colonies:</p> | ||
| + | <ul><li>Plate 1 (positive control pSB1A3 single cut, old ligase): ca. 150 colonies</li> | ||
| + | <li>Plate 2 (pSB1C3 + T7promoter, RBS, lacZ, ligation ratio I:V = 1): ca. 200 colonies</li> | ||
| + | <li>Plate 3 (pSB1C3 + T7promoter, RBS, lacZ, ligation ratio I:V = 0.4): ca. 30 colonies</li> | ||
| + | <li>Plate 4 (pSB1C3 + blue light sensor, RBS, lacZ, ligation ratio I:V = 1): ca. 1500 colonies</li> | ||
| + | <li>Plate 5 (pSB1C3 + blue light sensor, RBS, lacZ, ligation ratio I:V = 0.4): ca. 500 colonies</li> | ||
| + | <li>Plate 6 (vector background pSB1C3): 5 colonies</li> | ||
| + | <li>Plate 7 (positive control, pSB1A3 single cut, new ligase): ca. 200 colonies</li> | ||
| + | <li>Plate 8 (insert background blue light sensor, RBS, lacZ): ca. 50 colonies</li> | ||
| + | <li>Plate 9 (insert background T7promoter, RBS, lacZ): 3 colonies | ||
| + | </li></ul> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Cloning</span></h3> | ||
| + | <div class="cloning"> | ||
| + | <h4><span class="mw-headline" id="Picking_of_clones">Picking of clones</span></h4> | ||
| + | <p>3 colonies were picked each from plates 2, 3 (Part BBa_K568003 in pSB1C3) and 4, 5 (Part BBa_568002 in pSB1C3) and then inoculated in 5 ml LB Cam each. The cultures were incubated at 37°C over night for miniprep tomorrow.</p> | ||
| + | <p><br /></p> | ||
| + | <h4><span class="mw-headline" id="Cloning_of_Part_BBa_K568004">Cloning of Part BBa_K568004</span></h4> | ||
| + | <p>To ligate the synthetic construct (cut yesterday with E, S) with T7ptag in pSB1C3, the following digestion was done:</p> | ||
| + | <p><a href="/wiki/images/8/8e/Digest16092011.jpg" class="image" rel="lightbox"><img alt="Digest16092011" src="/wiki/images/8/8e/Digest16092011.jpg" width="400" /></a></p> | ||
| + | <p>After the digestion was completed, the sample was loaded onto a preparative 1 % agarose gel. After electrophoresis at 120 V, the correct bands were cut out on the transilluminator (365 nm, make sure UV lamp isn't switched on longer than absolutely necessary). Subsequently, the following gel photo was recorded:</p> | ||
| + | <p><a href="/wiki/images/9/94/160911_prep_digest_pSB1C3_t7ptag.jpg" class="image" rel="lightbox"><img alt="160911 prep digest pSB1C3 t7ptag" src="/wiki/images/9/94/160911_prep_digest_pSB1C3_t7ptag.jpg" width="400" /></a></p> | ||
| + | <p>pSB1C3 with T7ptag is 4743 bp long, which corresponds with the location of the cut-out band.</p> | ||
| + | <p>As a next step, the DNA was purified from the gel slice using the Promega Wizard Kit. The concentration of the product was determined to be 6 ng/µl using Nanodrop. This value is not reliable, but for lack of a second, quick way to measure the DNA concentration, the following ligation was perpared using the value nevertheless. Incubation of the Ligation occurred over night at 16 °C.</p> | ||
| + | <p><a href="/wiki/images/1/14/Ligation160911.jpg" class="image" rel="lightbox"><img alt="Ligation160911" src="/wiki/images/1/14/Ligation160911.jpg" width="400" /></a></p> | ||
| + | <p><br /> | ||
| + | The product of yesterday's ligation was purified using glycogen-ethanol-precipitation (see methods). The product was stored at -20°C for transformation tomorrow.</p> | ||
| + | </div> | ||
<h3> <span class="mw-headline">Other Work</span></h3> | <h3> <span class="mw-headline">Other Work</span></h3> | ||
<div class="otherwork"> | <div class="otherwork"> | ||
| - | <h4> <span class="mw-headline" id="Electrocompetent_cells_cp919">Electrocompetent cells cp919</span></h4> | + | <h4><span class="mw-headline" id="Electrocompetent_cells_cp919">Electrocompetent cells cp919</span></h4> |
<dl><dd>new electrocompetent cells (cp919) were made by the following protocol: (it had to be adjusted due to circumstances) | <dl><dd>new electrocompetent cells (cp919) were made by the following protocol: (it had to be adjusted due to circumstances) | ||
</dd></dl> | </dd></dl> | ||
| Line 815: | Line 890: | ||
</dd></dl> | </dd></dl> | ||
</dd></dl> | </dd></dl> | ||
| - | < | + | <h4><span class="mw-headline" id="New_media_and_plates">New media and plates</span></h4> |
| - | LB, LB for Agar plates (Cam and Amp) and Luria medium were prepared. Plates were prepared.</p> | + | <p>LB, LB for Agar plates (Cam and Amp) and Luria medium were prepared. Plates were prepared.</p> |
</div> | </div> | ||
| - | <h2> <span class="mw-headline" id="17-09-2011">17-09-2011</span></h2> | + | <h2><span class="mw-headline" id="17-09-2011">17-09-2011</span></h2> |
| - | <p><b>People: Simon, Anna</b></p><p><br /></p> | + | <p><b>People: Simon, Anna</b></p> |
| + | <h3> <span class="mw-headline">Cloning</span></h3> | ||
| + | <div class="cloning"> | ||
| + | <h4><span class="mw-headline" id="Minipreps_of_parts_BBa_K568003_and_BBa_K568002">Minipreps of parts BBa_K568003 and BBa_K568002</span></h4> | ||
| + | <p>Yesterday's overnight cultures were prepped today according to protocol. A total of 12 cultures was prepped, 6 of which contained pSB1C3 with K568003 while the other 6 contained pSB1C3 with K568002. | ||
| + | After the miniprep, the following concentrations were measured using Nanodrop:</p> | ||
| + | <p>Part K568003 (T7prom, RBS, lacZ):</p> | ||
| + | <ul><li> 2a: 86.5 ng/µl</li> | ||
| + | <li> 2b: 71.0 ng/µl</li> | ||
| + | <li> 2c: 63.5 ng/µl</li> | ||
| + | <li> 3a: 66.5 ng/µl</li> | ||
| + | <li> 3b: 88.5 ng/µl</li> | ||
| + | <li> 3c: 85.0 ng/µl | ||
| + | </li></ul> | ||
| + | <p>Part K568002 (blue light promoter, RBS, LacZ):</p> | ||
| + | <ul><li> 4a: 82.0 ng/µl | ||
| + | </li> | ||
| + | <li> 4b: 78.0 ng/µl</li> | ||
| + | <li> 4c: 80.0 ng/µl</li> | ||
| + | <li> 5a: 74.5 ng/µl</li> | ||
| + | <li> 5b: 97.0 ng/µl</li> | ||
| + | <li> 5c: 100 ng/µl | ||
| + | </li></ul> | ||
| + | <h4><span class="mw-headline" id="Restriction_digest_and_control_gel">Restriction digest and control gel</span></h4> | ||
| + | <p>To check whether the prepped DNA samples contained the correct parts, the following digestion was performed:</p> | ||
| + | <p><a href="/wiki/images/a/ab/17092011controldigest.jpg" class="image" rel="lightbox"><img alt="17092011controldigest" src="/wiki/images/a/ab/17092011controldigest.jpg" width="400" /></a></p> | ||
| + | <p>After the digest was completed, the samples were loaded onto a 0.8% agarose gel. After electrophoresis, the following gel photo was recorded:</p> | ||
| + | <p><a href="/wiki/images/6/60/170911_analytic_digests_pSB1C3_with_T7promLacZ_or_bluelightsensorLacZ.jpg" class="image" rel="lightbox"><img alt="170911 analytic digests pSB1C3 with T7promLacZ or bluelightsensorLacZ" src="/wiki/images/6/60/170911_analytic_digests_pSB1C3_with_T7promLacZ_or_bluelightsensorLacZ.jpg" width="400" /></a></p> | ||
| + | <p>All bands look fine. Samples 1-6 correspond to the minipreps of K568003. Here the insert is 3147 bp and the vector is 2070 bp long. The bands verify this. Samples 7 - 12 correspond to the minipreps of K568002, which has an insert of 3187 bp and the same 2070 bp vector. Again, the bands show that this is correct.</p> | ||
| + | <h4><span class="mw-headline" id="Glycogen.2Fethanol_precipitation">Glycogen/ethanol precipitation</span></h4> | ||
| + | <p>Yesterday's ligation was purified using glycogen/ethanol precipitation. The yielded DNA samples were used for transformation</p> | ||
| + | <h4><span class="mw-headline" id="Transformations">Transformations</span></h4> | ||
| + | <p>The transformations listed below were carried out. After electroporation the cells were incubated in 1 ml SOC medium for 1 h at 37 °C before plating.</p> | ||
| + | <p><b>For Redlight Sensor Characterization</b></p> | ||
| + | <p>All plasmids were electroporated into the new electrocompetent CP919 cells for characterization of the red light sensor later this week.</p> | ||
| + | <ul><li> Plate Number 1: Part K322127 in pSB1C3 (tube label "K322127 miniprep 04.09.2011"). Plated onto LB Cam plate.</li> | ||
| + | <li> Plate Number 2: Part K568000 (tube label "5.9.11, 2 vom 1.9, redlight in pSB1C3, 83 ng/µl"). This is our PCR product (redlight sensor). Plated onto LB Cam plate.</li> | ||
| + | <li> Plate Number 3: cph8 in pSB1A3 (tube label "cph8 Miniprep 4.9.11"). Plated onto both LB Amp. | ||
| + | </li></ul> | ||
| + | <p><br /> | ||
| + | <b>For generation of Part BBa_K568004</b></p> | ||
| + | <p>The purified products of yesterday's ligation were electroporated into DH5alpha and plated onto LB Cam plates.</p> | ||
| + | <ul><li> Plate Number 4: Ligation 1 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 5.</li> | ||
| + | <li> Plate Number 5: Ligation 2 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 10.</li> | ||
| + | <li> Plate Number 6: Ligation 3 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 15.</li> | ||
| + | <li> Plate Number 7: Ligation 4 (16-09-2011), vector background pSB1C3 T7ptag. | ||
| + | </li></ul> | ||
| + | <p>The purified products of the ligation performed on 15-09-2011 were electroporated into DH5alpha, just to make sure that the part is really contained in pSB1A3 and not pSB1C3. Therefore, the transformed cells were plate onto LB Amp and LB Cam plates. The plate Number refers to the LB Amp plate as well as the LB Cam plate in all cases.</p> | ||
| + | <ul><li> Plate Number 8: Ligation 1 (15-09-2011), positive control pSB1A3 single cut</li> | ||
| + | <li> Plate Number 9: Ligation 2 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 5.</li> | ||
| + | <li> Plate Number 10: Ligation 3 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 10. | ||
| + | </li> | ||
| + | <li> Plate Number 11: Ligation 4 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 15.</li> | ||
| + | <li> Plate Number 12: Ligation 5 (15-09-2011) vector background pSB1A3 T7ptag (purify 1.9.) | ||
| + | </li></ul> | ||
| + | <p><b>For cloning of I746907 into pSB6A1</b></p> | ||
| + | <p>We want to use I746907 as a reporter part for our optogenetical AND-gate in e. coli CP919. Therefore the part must be transformed into a low copy vector, since the AND-gate is in pSB1C3 which is a high copy vector. Because CP919 already has a Kan-Resistance, the low copy vector used for this needs to carry Amp-Resistance. That's why we chose pSB6A1, a low copy vector with Amp-Resistance. | ||
| + | The DNA was gained from 2011 Kit Plate 1 well 1K by resuspension in 10 µl ddH2O. This was then used for electroporation into DH5alpha which were plated onto LB Amp plates after transformation.</p> | ||
| + | <ul><li> Plate Number 13: pSB6A1, 0.5 µl DNA used for electroporation</li> | ||
| + | <li> Plate Number 14: pSB6A1, 1.0 µl DNA used for electroporation</li> | ||
| + | <li> Plate Number 15: pSB6A1, 8.5 µl DNA used for electroporation | ||
| + | </li></ul> | ||
| + | <p><br /> | ||
| + | <b>For Testing of I746907</b></p> | ||
| + | <p>To be able to characterize part I746907, we used a chemical transformation into BL21 DE3 (see methods). 1.5 µl of the DNA were used (c = 63 ng/µl). The transformed cells were plated onto a LB Amp plate.</p> | ||
| + | <ul><li> Plate Number 16: part I746907 in pSB1A2 | ||
| + | </li></ul> | ||
| + | </div> | ||
<h3> <span class="mw-headline">Results</span></h3> | <h3> <span class="mw-headline">Results</span></h3> | ||
<div class="results"> | <div class="results"> | ||
| - | <h4> | + | <h4><span class="mw-headline" id="sequencing_results_2">sequencing results</span></h4> |
| - | <p> | + | <p>(Susan) |
| - | compared to our parts designed in registry and pSB1C3:</p><p | + | compared to our parts designed in registry and pSB1C3:</p> |
| + | <p>pSB1C3_opt_and-gate_110913_clone_2a_VR-GATC-VR-547731: no results, bad sequencing quality.</p> | ||
| + | <p>pSB1C3_opt_and-gate_110913_clone_6b_VR-GATC-VR-547731: part ok! End of part and plasmid backbone were sequenced.</p> | ||
| + | <p>pSB1C3_synth_construct_110910_clone_5a_VF2-GATC-VF2-545457: Part ok! inserted in backbone</p> | ||
| + | <p>pSB1C3_synth_construct_110910_clone_6b_VF2-GATC-VF2-545457: Part ok! inserted in backbone, 8 bases could not be detected, so part 5a is better, here the whole sequence is right. After looking at the raw data, sequence seems to be ok.</p> | ||
</div> | </div> | ||
| - | <h2> <span class="mw-headline" id="18-09-2011">18-09-2011</span></h2> | + | <h2><span class="mw-headline" id="18-09-2011">18-09-2011</span></h2> |
<p><b>People: Simon, Flo</b></p> | <p><b>People: Simon, Flo</b></p> | ||
| + | <h3> <span class="mw-headline">Results</span></h3> | ||
| + | <div class="results"> | ||
| + | <p>The transformations done yesterday worked quite well.</p> | ||
| + | <ul><li> Plate 1 (Cam): lawn with a few single colonies</li> | ||
| + | <li> Plate 2 (Cam): lawn with a few single colonies</li> | ||
| + | <li> Plate 3 (Amp): ca. 20 colonies</li> | ||
| + | <li> Plate 4 (Cam): ca. 300 colonies</li> | ||
| + | <li> Plate 5 (Cam): ca. 40 colonies</li> | ||
| + | <li> Plate 6 (Cam): ca. 20 colonies</li> | ||
| + | <li> Plate 7 (Cam): 4 colonies | ||
| + | </li></ul> | ||
| + | <p>No colonies grew on plates 8 through 12 with Cam. This means that the part was really cloned into pSB1A3. Hence, we use colonies from plates 4 - 6 for miniprepping of part BBa_K568004 in pSB1C3.</p> | ||
| + | <ul><li> Plate 8 (Amp): > 1000 colonies</li> | ||
| + | <li> Plate 9 (Amp): no colonies</li> | ||
| + | <li> Plate 10 (Amp): ca. 900 colonies</li> | ||
| + | <li> Plate 11 (Amp): ca. 900 colonies</li> | ||
| + | <li> Plate 12 (Amp): ca. 30 colonies</li> | ||
| + | <li> Plate 13 (Amp): ca. 50 colonies</li> | ||
| + | <li> Plate 14 (Amp): ca. 80 colonies | ||
| + | </li> | ||
| + | <li> Plate 15 (Amp): ca. 120 colonies</li> | ||
| + | <li> Plate 16 (Amp): ca. 300 colonies | ||
| + | </li></ul> | ||
| + | </div> | ||
<h3> <span class="mw-headline">Cloning</span></h3> | <h3> <span class="mw-headline">Cloning</span></h3> | ||
<div class="cloning"> | <div class="cloning"> | ||
| - | <h4> <span class="mw-headline" id="Picking_of_clones_of_Bba_K568004_in_pSB1C3_for_miniprep">Picking of clones of Bba_K568004 in pSB1C3 for miniprep</span></h4> | + | <h4><span class="mw-headline" id="Picking_of_clones_of_Bba_K568004_in_pSB1C3_for_miniprep">Picking of clones of Bba_K568004 in pSB1C3 for miniprep</span></h4> |
| - | <p>3 clones each were picked from plates 4, 5 and 6 (transformation from 17-09-2011) and 5 ml LB + Cam were inoculated and vortexed shortly for equal distribution of bacterial cells.</p><p>Falcon tubes were set almost horizontally in the 37°C-incubator to maximize oxygen input and shaking occurred @ 250 rpm for 3 h and 180 rpm for another 4.5 h.</p> | + | <p>3 clones each were picked from plates 4, 5 and 6 (transformation from 17-09-2011) and 5 ml LB + Cam were inoculated and vortexed shortly for equal distribution of bacterial cells.</p> |
| - | <h4> <span class="mw-headline" id="Miniprep_of_3_x_3_clones_of_Bba_K568004_in_pSB1C3">Miniprep of 3 x 3 clones of Bba_K568004 in pSB1C3</span></h4> | + | <p>Falcon tubes were set almost horizontally in the 37°C-incubator to maximize oxygen input and shaking occurred @ 250 rpm for 3 h and 180 rpm for another 4.5 h.</p> |
| - | <p>Miniprep done according to kit instruction (Metabion "mi-plasmid mini prep" | + | <h4><span class="mw-headline" id="Miniprep_of_3_x_3_clones_of_Bba_K568004_in_pSB1C3">Miniprep of 3 x 3 clones of Bba_K568004 in pSB1C3</span></h4> |
| + | <p>Miniprep done according to kit instruction (Metabion "mi-plasmid mini prep") [See Methods]</p> | ||
<p><i>Changes:</i> </p> | <p><i>Changes:</i> </p> | ||
| - | <ul><li>All 5 ml bacterial suspension were used for the prep | + | <ul><li>All 5 ml bacterial suspension were used for the prep</li> |
| - | </li><li>Autoclaved water for elution was pre-warmed to ~60°C | + | <li>Autoclaved water for elution was pre-warmed to ~60°C |
| + | </li></ul> | ||
| + | <p><i>Names (according to plate # from 17-09-2011):</i></p> | ||
| + | <ul><li> K568004_<b>4-1</b>, c = 64.0 ng/µl </li> | ||
| + | <li> K568004_<b>4-2</b>, c = 92.0 ng/µl</li> | ||
| + | <li> K568004_<b>4-3</b>, c = 62.0 ng/µl</li> | ||
| + | <li> K568004_<b>5-1</b>, c = 67.0 ng/µl | ||
| + | </li> | ||
| + | <li> K568004_<b>5-2</b>, c = 76.5 ng/µl</li> | ||
| + | <li> K568004_<b>5-3</b>, c = 74.5 ng/µl </li> | ||
| + | <li> K568004_<b>6-1</b>, c = 75.5 ng/µl</li> | ||
| + | <li> K568004_<b>6-2</b>, c = 61.5 ng/µl</li> | ||
| + | <li> K568004_<b>6-3</b>, c = 74.5 ng/µl | ||
</li></ul> | </li></ul> | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
</div> | </div> | ||
<h3> <span class="mw-headline">Other work</span></h3> | <h3> <span class="mw-headline">Other work</span></h3> | ||
<div class="otherwork"> | <div class="otherwork"> | ||
| - | <h4> <span class="mw-headline" id="Preparation_of_iGEM-parts_for_shipping">Preparation of iGEM-parts for shipping</span></h4> | + | <h4><span class="mw-headline" id="Picking_of_clones_2">Picking of clones</span></h4> |
| - | <p> | + | <p>A clone of plate 16 (BL21 DE3 with I746907) was picked and inoculated in 5 ml LB Amp for testing tomorrow.</p> |
| + | <p>2 clones were picked each from plates 12 - 15 (DH5alpha containing pSB6A1) and inoculated in 5 ml Amp for miniprep tomorrow.</p> | ||
| + | <h4><span class="mw-headline" id="Preparation_of_iGEM-parts_for_shipping">Preparation of iGEM-parts for shipping</span></h4> | ||
| + | <p><a href="/wiki/images/2/25/Screen_shot_2011-09-19_at_8.20.43_PM.png" class="image" rel="lightbox"><img alt="Screen shot 2011-09-19 at 8.20.43 PM.png" src="/wiki/images/2/25/Screen_shot_2011-09-19_at_8.20.43_PM.png" width="400" /></a></p> | ||
| + | </div> | ||
| + | |||
| + | <h2><span class="mw-headline" id="19-09-2011">19-09-2011</span></h2> | ||
| + | <p><b>People: Simon, Anna</b></p> | ||
| + | <p>Our Biobricks will be picked up by FedEx today between 1 pm and 3 pm for shipping to the registry. 6 out of 7 Biobricks are ready for take-off, but BBa_K568004 has to be finished in time! </p> | ||
| + | <h3> <span class="mw-headline">Cloning</span></h3> | ||
| + | <div class="cloning"> | ||
| + | <h4><span class="mw-headline" id="Part_BBa_K568004">Part BBa_K568004</span></h4> | ||
| + | <p>The 9 prepped samples of BBa_K568004 have to be checked for correct sizes of vector and insert. The following digestion was prepared:</p> | ||
| + | <p><br /> | ||
| + | <a href="/wiki/images/2/20/19092011controldigest.jpg" class="image" rel="lightbox"><img alt="19092011controldigest" src="/wiki/images/2/20/19092011controldigest.jpg" width="400" /></a></p> | ||
| + | <p><br /> | ||
| + | Due to time reasons, 10 µl of the sample was digested for 1 min in the microwave at 800 W, while the other 10 µl of the sample were incubated at 37 °C.</p> | ||
| + | <p>The product of quick digest in the microwave was separated using electrophoresis with an analytical 0.8 % agarose gel. This yielded the following picture:</p> | ||
| + | <p><br /></p> | ||
| + | <p><a href="/wiki/images/c/c7/19092011controldigestshort.jpg" class="image" rel="lightbox"><img alt="19092011controldigestshort" src="/wiki/images/c/c7/19092011controldigestshort.jpg" width="400" /></a></p> | ||
| + | <p><br /></p> | ||
| + | <p>The bands of all samples look fine. The insert BBa_K568004 is 3069 bp long, the pSB1C3 vector is 2070 bp long. All samples show these bands. We chose sample 8 for shipping, because it looked "cleanest" on the gel.</p> | ||
| + | <p><br /> | ||
| + | The product of the "normally" incubated restriction digest (1 h at 37 °C) was also loaded onto an analytical 0.8 % agarose gel. The subsequent electrophoresis resulted in the following picture:</p> | ||
| + | <p><br /> | ||
| + | <a href="/wiki/images/6/60/19092011_control_long_BBa_K568004.jpg" class="image" rel="lightbox"><img alt="19092011 control long BBa K568004" src="/wiki/images/6/60/19092011_control_long_BBa_K568004.jpg" width="400" /></a></p> | ||
| + | <p><br /> | ||
| + | Again, the bands are at the expected positions.</p> | ||
| + | <p><br /></p> | ||
| + | <h4><span class="mw-headline" id="Miniprep_of_pSB6A1">Miniprep of pSB6A1</span></h4> | ||
| + | <p>All six overnight cultures of pSB6A1 in DH5alpha were miniprepped according to metabion's protocol. The cell pellets were pink, indicating that the cells contained the desired plasmid, which carries an RFP-coding sequence as a standard insert. Due to a mistake, the pellet of one sample was lost.</p> | ||
| + | <p>The concentrations were measured using Nanodrop:</p> | ||
| + | <ul><li> Sample 1: c = 93.0 ng/µl</li> | ||
| + | <li> Sample 2: c = 79.0 ng/µl</li> | ||
| + | <li> Sample 3: c = 77.5 ng/µl</li> | ||
| + | <li> Sample 4: c = 86.5 ng/µl</li> | ||
| + | <li> Sample 5: c = 102 ng/µl | ||
| + | </li></ul> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Other Work</span></h3> | ||
| + | <div class="otherwork"> | ||
| + | <p>We prepared the miniprepped pSB1C3 with K568004 for shipping. For used amount see the table found in labbook on 18-09-2011.</p> | ||
| + | <p>Preparations for testing of red light sensor tomorrow:</p> | ||
| + | <p>1 clone was picked from each of the following plates (all plates from transformations performed on 17-09-2011):</p> | ||
| + | <ul><li> Plate 1 (K322127), inoculated in 5 ml LB Cam</li> | ||
| + | <li> Plate 2 (K568000), inoculated in 5 ml LB Cam</li> | ||
| + | <li> Plate 3 (cph8), inoculated in 5 ml LB Amp | ||
| + | </li></ul> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Testing</span></h3> | ||
| + | <div class="testing"> | ||
| + | <h4><span class="mw-headline" id="GFP_Assay">GFP Assay</span></h4> | ||
| + | <p>200 µl of the overnight culture of BL21 (DE3) with I746907 (T7 promotor with GFP) in pSB1A2 were inoculated into 5 ml of fresh LB Amp and incubated for 1.5 hours at 37 °C. | ||
| + | Before the samples for measurement in the plate reader were prepared, we made a change of medium to M63, because LB shows slight fluorescence itself.</p> | ||
| + | <p><a href="/wiki/images/0/05/GFP_Assay.jpg" class="image" rel="lightbox"><img alt="GFP Assay" src="/wiki/images/0/05/GFP_Assay.jpg" width="400" /></a></p> | ||
| + | <p>The measurement using the plate reader occured over night, with heating the plate to 37 °C and shaking it between measurements.</p> | ||
| + | <p>Used filters: | ||
| + | excitation filter: 485 nm, emission filter: 520 nm</p> | ||
| + | <p><b>Problem</b>: Fluorescence was measured every 10 minutes while measurement of Absorbance occured only every 100 minutes. This led to spikes in the diagram.</p> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Results</span></h3> | ||
| + | <div class="results"> | ||
| + | <p>no concentration dependency of GFP induction (between 1.5 and 0.1 mM IPTG), as IPTG concentration was in saturation range</p> | ||
| + | <p>this table shows measured values with</p> | ||
| + | <ul><li> calculated: Fluorescence/Absorbance</li> | ||
| + | <li> mean of triplicate measurement</li> | ||
| + | <li> adjusted to 100 %</li> | ||
| + | <li> corrected for outliers</li> | ||
| + | <li> shortened to 700 min | ||
| + | </li></ul> | ||
| + | <p><br /></p> | ||
| + | <p><br /> | ||
| + | <a href="/wiki/images/5/58/2011-09-19_GFP_Assay_Tabelle_prozentual.jpg" class="image" rel="lightbox"><img alt="2011-09-19 GFP Assay Tabelle prozentual" src="/wiki/images/5/58/2011-09-19_GFP_Assay_Tabelle_prozentual.jpg" width="400" /></a></p> | ||
| + | <p><a href="/wiki/images/d/d5/2011-09-19_GFP_Assay_Diagramm_prozentual.jpg" class="image" rel="lightbox"><img alt="2011-09-19 GFP Assay Diagramm prozentual" src="/wiki/images/d/d5/2011-09-19_GFP_Assay_Diagramm_prozentual.jpg" width="400" /></a></p> | ||
| + | <p><br /></p> | ||
| + | <p>ideas for new GFP Assay:</p> | ||
| + | <ul><li> much lower IPTG concentrations</li> | ||
| + | <li> measurement of Absorbance every ten minutes | ||
| + | </li></ul> | ||
| + | </div> | ||
| + | |||
| + | <h2><span class="mw-headline" id="20-09-2011">20-09-2011</span></h2> | ||
| + | <p><b>People: Simon, Anna</b></p> | ||
| + | <h3> <span class="mw-headline">Cloning</span></h3> | ||
| + | <div class="cloning"> | ||
| + | <p>To be able to test the optogenetical AND-Gate in CP919, we need to transform the desired reporter part into a low copy vector. | ||
| + | Therefore, BBa_ I746907 will be ligated into pSB6A1.</p> | ||
| + | <h4><span class="mw-headline" id="Digest_2">Digest</span></h4> | ||
| + | <p>The following digest was performed:</p> | ||
| + | <p><br /> | ||
| + | <a href="/wiki/images/9/9a/Digest20092011.jpg" class="image" rel="lightbox"><img alt="Digest20092011" src="/wiki/images/9/9a/Digest20092011.jpg" width="400" /></a></p> | ||
| + | <p><br /> | ||
| + | After this, the samples were separated by electrophoresis using a preparative 1 % agarose gel. The correct gel bands were cut out on the transilluminator at 365 nm (keep irradiation time as short as possible). After this, the following photo was recorded:</p> | ||
| + | <p><br /> | ||
| + | <a href="/wiki/images/8/8a/200911_prep_digest_pSB6A1_and_I746907.jpg" class="image" rel="lightbox"><img alt="200911 prep digest pSB6A1 and I746907" src="/wiki/images/8/8a/200911_prep_digest_pSB6A1_and_I746907.jpg" width="400" /></a></p> | ||
| + | <p><br /> | ||
| + | The bands match the expectations. </p> | ||
| + | <p>Samples - : The cut out band around 4 kb is the cut vector pSB6A1, which is 4022 bp long. The band visible at around 1 kb is the RFP coding sequence (1069 bp).</p> | ||
| + | <p>Samples - : Part I746907 is 924 bp long. This corresponds to the cut out bands. The vector pSB1A2 can be seen at 2 kb (it is 2079 bp long).</p> | ||
| + | <h4><span class="mw-headline" id="Ligation_5">Ligation</span></h4> | ||
| + | <p>After purification of the DNA from the gel slices using the Promega Wizard Kit, the following ligation was prepared and incubated at 16 °C over night:</p> | ||
| + | <p><a href="/wiki/images/2/2f/Ligation200911.jpg" class="image" rel="lightbox"><img alt="Ligation200911" src="/wiki/images/2/2f/Ligation200911.jpg" width="400" /></a></p> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Testing</span></h3> | ||
| + | <div class="testing"> | ||
| + | <h4><span class="mw-headline" id="Miller_Assay_for_testing_red_light_sensors">Miller Assay for testing red light sensors</span></h4> | ||
| + | <p>Comparision of the parts:</p> | ||
| + | <ul><li> our red light sensor K568000, the PCR product of K322127, pSB1C3 (C) in cp919</li> | ||
| + | <li> K322127 of the team Edinburgh 2010, pSB1C3 (C) in cp919</li> | ||
| + | <li> cpH8 in pSB1A3 in cp919 as a negative control | ||
| + | </li></ul> | ||
| + | <p><br /> | ||
| + | three lighting options:</p> | ||
| + | <ul><li> off light (710 nm)</li> | ||
| + | <li> on light (626 nm)</li> | ||
| + | <li> dark | ||
| + | </li></ul> | ||
| + | <p><br /> | ||
| + | light sensitive cultures were grown in the dark for five hours prior to the experiment</p> | ||
| + | <p>OD600 around 2.5 (of cultures before dilution at the start of the experiment), dilution in LB medium to about OD = 0.1 </p> | ||
| + | <p>Start of Assay: | ||
| + | - measurement of Abs(600nm)</p> | ||
| + | <p>- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood) <b>+ 420 µl LB Medium</b> + <b>20 µl sample</b></p> | ||
| + | <p>- 0,1 ml of the samples are taken and transferred to the Zbuffer</p> | ||
| + | <p>- mix the solution by vortexing for 10 s (all samples equal vortexing time)</p> | ||
| + | <p>- <b>cenrrifugation: 1 min, RT, 4000 rcf</b></p> | ||
| + | <p>- transferring of 100 μl supernatant to 96 well plate (for photometer)</p> | ||
| + | <p>- initiation of assay with 20 μl of ONPG (4mg/ml) NOTE START TIME</p> | ||
| + | <p>- incubation at 37 °C</p> | ||
| + | <p>- stop reaction with 50 μl of 1 M Na2CO3 NOTE STOP TIME </p> | ||
| + | <p>- measurement of Abs (420nm) and Abs (550nm)</p> | ||
| + | <p><br /></p> | ||
| + | <p><a href="/wiki/images/7/7c/2011-09-20_MillerAssay_Tabelle.jpg" class="image" rel="lightbox"><img alt="2011-09-20 MillerAssay Tabelle" src="/wiki/images/7/7c/2011-09-20_MillerAssay_Tabelle.jpg" width="400" /></a></p> | ||
| + | <p><a href="/wiki/images/0/0f/2011-09-20_MillerAssay_Diagramm.jpg" class="image" rel="lightbox"><img alt="2011-09-20 MillerAssay Diagramm" src="/wiki/images/0/0f/2011-09-20_MillerAssay_Diagramm.jpg" width="400" /></a></p> | ||
| + | </div> | ||
| + | <h3> <span class="mw-headline">Results</span></h3> | ||
| + | <div class="results"> | ||
| + | <p><b>In none of the cells with either the K568000 or the K322127 lacZ could be detected.</b></p> | ||
| + | <p>possible improvements for further experiments:</p> | ||
| + | <ul><li> make sure, the cells are in log phase, diluting down earlier, e.g. at a OD600 of about 0.7, however cells shouldn't be in lag phase after 210 min, so at least the last values represent lacZ expression in log phase | ||
| + | </li></ul> | ||
| + | </div> | ||
| + | |||
| + | <h2><span class="mw-headline" id="21-09-2011">21-09-2011</span></h2> | ||
| + | <p>Wiki freeze day! </p> | ||
| + | <p>Yesterday's ligation was incubated at 65 °C for 10 minutes to heat inactivate the enzymes. The samples were stored at -20 °C and can be used for transformation, as soon as we get to characterizing our optogenetical AND-Gate (BBa_568001). We still hope to generate some data to characterize this part in the near future...</p> | ||
</div> | </div> | ||
Latest revision as of 01:54, 22 September 2011
Cloning Part III + Results
People: Alex, Simon, Bea, Thorsten, Anna
24-08-2011
People: Alex, Simon, Bea, Thorsten
Cloning
Ligation
Three ligations in total were conducted. K238013 (S,P) was quick-ligated with lacZ (X,P), rbs (S,P) with T7Pol (X,P) and T7Prom (S,P) with lacZ (X,P).
Transformation
The ligations were transformed into DH5alpha. Additionally, K238013 (from23-08-2011) was transformed into DH5alpha. The ligation of K238013 with lacZ is plated onto Amp and Cm, rbs with T7pol is plated onto Amp, T7Prom with lacZ onto Kan and K238013 is plated onto Amp and Cm.
Testing
The two cultures of K322127 in DH5alpha incubated over night with red light (ca. 630 nm) were split. One half was further incubated with red light, while the other half was incubated with sun light and light from a table-lamp over night.
Test of negative controls on S-Gal: T7prom (Kan) transformed into DH5alpha and K322127 (Cm) transformed into BL21 were inoculated onto S-Gal plates with the respective antibiotics. The are incubated over night, and should not yield black colonies, as they both should not be able to express functional lacZ.
Results
Expression test of reporter plasmid: The cultures with ligation of T7prom and lacZ transformed into BL21 and induced with IPTG showed black colonies on the S-Gal plates. However, the cultures, which were NOT induced also showed black colonies of the same intensity. At the current point, we cannot sufficiently explain this.
Red light sensor assembly (Bea and Thorsten)
aim:
Ligation of red light sensor didn't work, yet. Therefore we ligated PCR product of BBa_K322127(X and P cut) into pSB3C5(S and P cut) to verify correct sites. Subsequent amplification and recutting of this part should lead to correct ligation with SupD.
In parallel, PCR was performed again in case of failed ligations with existent PCR-products.
Materials:
digest:
pSB3C5: S and P cut by Alex on 23.08.11, CAM resistance, 2,7 kbp vector backbone
PCR product of BBa_K322127 with primers BBa_K568000, derived from Biomers, sequence see below, 3,8 kbp (Florian 6.7.11 labelled with "K322127 PCR product aus gel ausgeschnitten")
pcr:
Template: BBa_K322127 ("Edinburgh part") conc: 150 ng/µl
primers (synthesized by biomers, 29.6.11, stock: 100µM):
- BBa_K568000_fwd: tatatctatatcgaattcgcggccgcttctagagtttacggctagctcagtcctaggta (59bp)
- BBa_K568000_rev: cgtgccggcggctgcagcggccgctactagtaagtccattctccccaaaaatg (53bp)
Procedure
digest:
10 µl PCR Product was cut with 1 µl XbaI and 1 µl PstI in 5 µl NEB4-buffer (total volume 50 µl) for 2h at 37°C with subsequent heat inactivation at 80°C for 20 minutes. Cut PCR-product was loaded on a 1% Agarose Gel in TAE buffer and run at 120 V for 1 h.
PCR:
amplification with Taq PCR Kit (NEB) according to Florian at 5.7.11. No water control was run due to lacking remaining buffer volume.
- 5 µl 10x standard buffer
- 1 µl dNTP's
- 0,25 µl Tag DNA polymerase
- 0,5 µl primers, respectively
- 0,4 µl Template DNA
- add up to 50 µl with sterile water
PCR-program:
- see 5.7.11
Bands were cut from Gel and DNA was isolated using Squeeze N Freeze (protocol see Methods)
Results
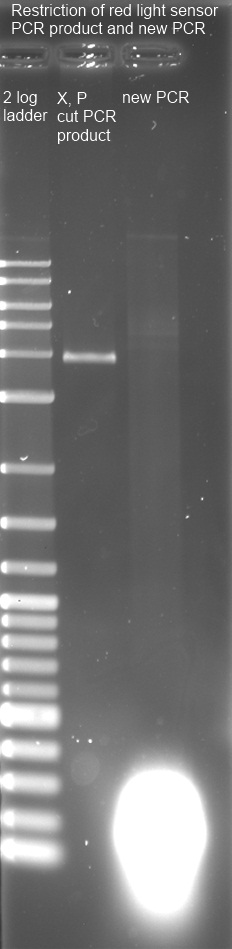
1% Agarose Gel of new PCR-product (line 3) and cut PCR-product (line2) with 2 log ladder (line 1)
cut PCR Fragment with correct size (3,8 kbp) was frozen after isolation. PCR yielded some short products with variable length. Used PCR-Kit is not suitable for amplification of long fragments. Experiment will be repeated tomorrow with correct Kit.
25-08-2011
People: Alex, Simon, Bea, Thorsten
Cloning
Digestion
R2 (red writing: MiniPrep iGEM R2 31.5.11) was digested with X,P and T7prom (black writing: 17.6.11 T7) with S,P. After heat-inactivation, GLP was added to the digested DNA. The samples were applied onto a 1 % Agarose gel. The samples, along with those from the PCR (see below), were applied as follows: 2lod DNA ladder (5 ul); R2 (20 ul); R2 (20 ul); T7prom (20 ul); T7 prom (20 ul); PCR cut; positive control; PCR approach 1; PCR approach 1; PCR approach 2; PCR approach 2; PCR approaches 1 and 2 pooled. The gel was run for 1:30 h at 110 V. The following bands were expected: 3.1 kbp for lacZ with rbs and 2 kbp for vector pSB1A2. 3.4 kbp for T7prom in pSB1AK8. The bands are excised from the gel and the DNA is recovered using freeze 'n squeeze.
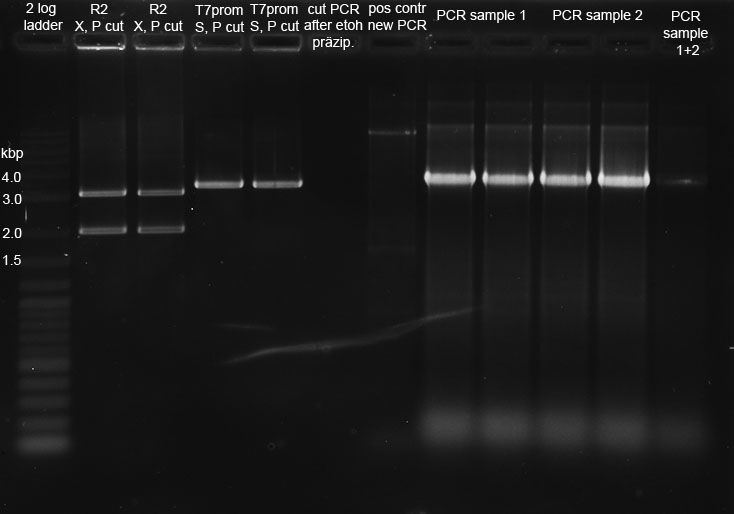
The original file before cutting of the bands seems to be corrupt.
Ligation
In the next step, K238013 (from 23-08-2011; cut with S,P) was ligated with rbs&lacZ (=R2; X,P), rbs (from 22-08-2011; S,P) with T7pol (from 23-08-2011; X,P) and T7prom (S,P) with rbs&lacZ (X,P). Each time, ligation was performed as normal and as quick-ligation. After that, the transformed cells were plated onto LB (Amp) plates and incubated over night.
New PCR of K322128 using Phusion High-Fidelity PCR-Kit (NEB/Finnzymes)
Material
BBa_K322127 with primers BBa_K568000, derived from Biomers, 3,8 kbp cut vector pSB1A2 digestion with X,P from Alex on 25.8.2011 (Florian 6.7.11 labelled with "K322127 PCR product aus gel ausgeschnitten")
Procedure
using same protocol as Florian on 11.7
PCR-Program:
1) Initial Denaturation: 98°C, 4'
2) 30 cycles of:
Denat.: 98°C, 30 s Annealing: 60°C, 15 s Extension: 72°C, 2´ 30 s
3) Final extension: 72°C, 8'
4) Hold: 4°C, oo
samples
only 2 samples were prepared using each:
35,5µl of ddH2O 10 µl5x buffer 1 µl dNTPS 1 µl of template DNA K322127 (1,5ng/µl ) 1 µl of Primer fwd (10µM) 1 µl of Primer rev (10µM)
only the 10kB positive control was done:
34µl of ddH2O 10 µl5x buffer 1 µl dNTPS 1 µl of template DNA (template for positive control) 2,5 µl of Primer (10kB-Mix)
after pcr the samples were applied on a 1% Agarose-gel
The bands are excised from the gel and the DNA is recovered using freeze 'n squeeze.
two times 10µl of the PCR product were digested with X,P
the digestion and the pcr product were stored at -20°C
Ethanol precipitation of digested PCR product
Material: PCR product cut with X,P from the 24.8.2011 protocol see Methods the sample was applied on an agarose gel because the nanodrop indicated no DNA
Result
The gel showed that no DNA was yielded (see picture Restriction & Ligation)
Repeated Digest and Ligation of PCR Product
Material: PCR product of BBa_K322127 with primers BBa_K568000, derived from Biomers, 3,8 kbp cut vector pSB1A2 digestion with X,P from Alex on 25.8.2011 (Florian 6.7.11 labelled with "K322127 PCR product aus gel ausgeschnitten") Procedure: The PCR product was digested with X,P. After heat-inactivation, GLP was added to the digested DNA. The samples were applied onto a 0,8 % Agarose gel. The gel was run for 1:30 h at 110 V. The bands are excised from the gel and the DNA is recovered using freeze 'n squeeze. Ligation of the cut fragment with the vector pSB1A2 (cut with X,P; see Restriction & Ligation above) using quick ligation protocol see Methods
Result
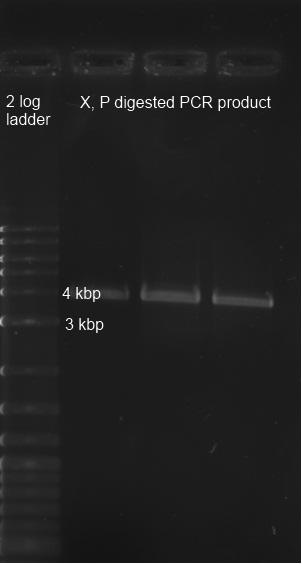
Testing
SDS-PAGE
1 OD_600 each from the K322127 in DH5alpha cultures from yesterday was taken. The OD_600 were: 0.835 for the first colony exposed to bright lamp (bright 1); 0.84 for the second colony exposed to bright lamp (bright 2); 1.685 for the first colony incubated with red light (red 1); 1.855 for the second colony incubated with red light (red 2). The cells were centrifuged at 13000 rcf for 8 min. After resuspension in 100 ul loading buffer, the tubes were incubated for 10 min at 95 °C. After that, the samples (5 ul and 10 ul) were loaded onto a 10 % SDS-PAGE with stacking gel. Pageruler was used as protein ladder. The gel was run for 2:00 h at 80 V, which was increased to 120 V when the buffer front reaching the separation gel. After that, the gel was stained with Coomassie blue and subsequently destained.
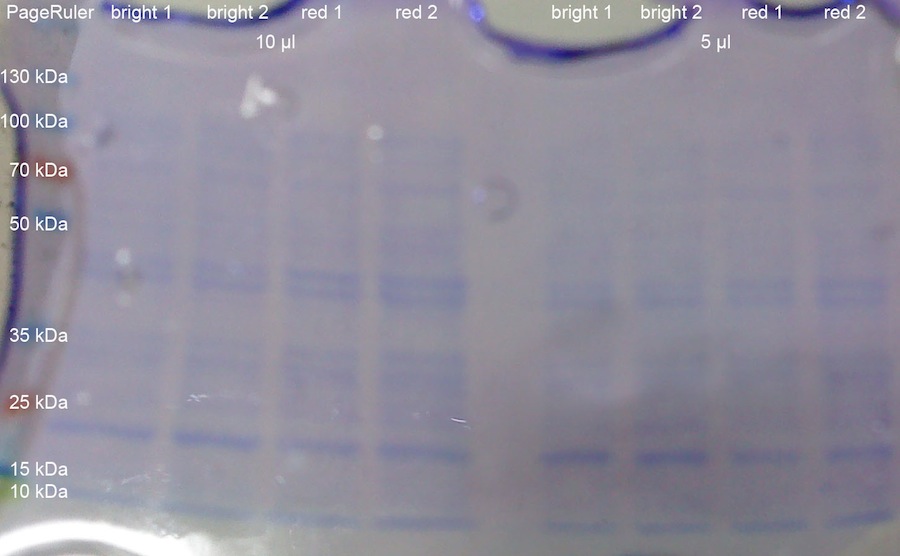
The gel does not show elevated intensities of the band at around 10 kDa for lacZ alpha peptide, which would have been expected.
T7 promoter in vitro assay
As proposed yesterday, a second in vitro assay was performed to find out if the plasmids contain a T7 promoter. This time, a third sample was made for each plasmid (same plasmids as yesterday, see above). In this sample (labeled "#xi") the RNase that we assume to be present were inhibited using RNA secure prior to addition of T7 polymerase and incubation. See methods for detailed procedure. The 2% agarose gel was run at 110 V, 400 mA for 1:30 hours.
Other Work
K322127 Inoculation & Incubation:
The following inoculations were performed:
- "K238013 in DH5 alpha", plate date: 24.08.2011, yesterday's transformation, in LB Amp
- "BM28 from Jeanette Winter", plate date: 31.05.2011, in LB Amp, Kan
- "green light 2", plate date: 27.6.2011, in LB Cm
- "green light 3", plate date: 27.6.2011, in LB Cm
- "green light 4", plate date: 27.6.2011, in LB Cm
- "K322127 in DH5 alpha, Kolonie 1, Licht-induziert", plate date: 22.08.11, in LB cm (the picked colonies were the black ones after over night incubation in light)
- "K322127 in DH5 alpha, Kolonie 2, Rotlicht-deaktiviert", plate date: 22.08.11, in LB cm (the picked colonies were white ones after over night incubation in red light)
Incubation over night at 37 °C, 200 rpm.
Results
The PCR was successful (see picture Restriction & Ligation), the positive control and both samples yielded DNA that is located at the right spot according to their length positive control (10kB) samples (3,8kB)
Results
24.08.11
T7 promoter in vitro assay
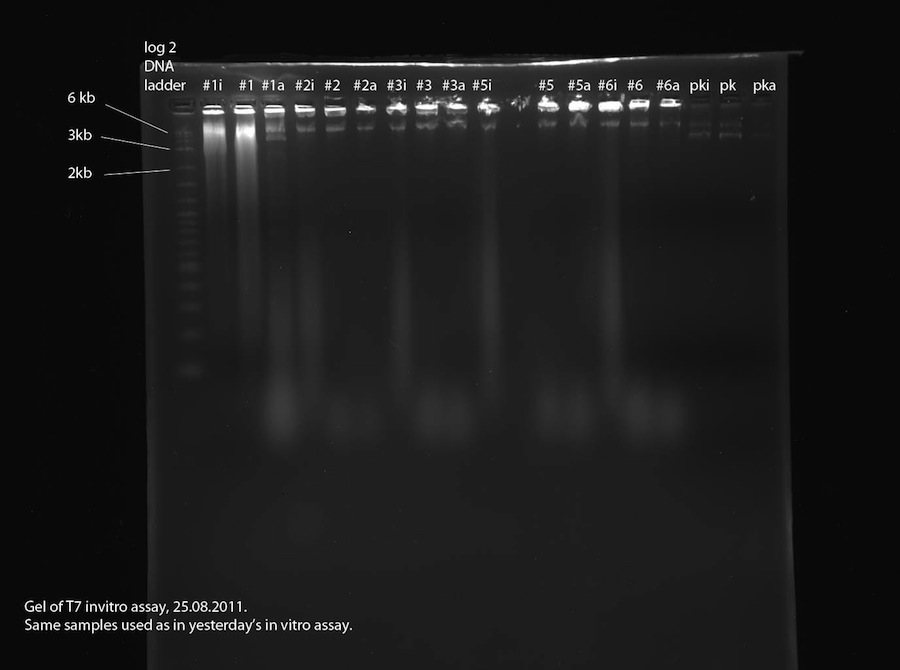
The samples labeled "#xi" were treated with RNA secure to inhibit potentially present RNase. The gel photo shows blurry signals in all of these lanes which aren't visible in lanes "#x" and "#xa". This proves that RNase is present in the DNA samples used in the assay. Also, the fact that the RNA signals are quite large and strong shows leads us to the conclusion that the examined plasmids all contain a T7 promoter. Sample 1 contains two plasmids (about 6.6 kb and 3.5 kb) which is the reason for the bad sequencing data. Samples 2, 3, 5 and 6 contain one 6.6 kb plasmid each. This should be our correct reporter plasmid, part 2b, containing T7 promoter and lacZ. To verify this, a sample shall be sequenced.
S-Gal negative Control:
DH5alpha cells transformed with T7prom yielded white colonies on the S-Gal plates. However, BL21 cells transformed with K322127 yielded all black colonies. Most likely, our strain of BL21 is unsuited for expression tests using S-Gal (might constitutively express lacZ).
Expression test of K322127
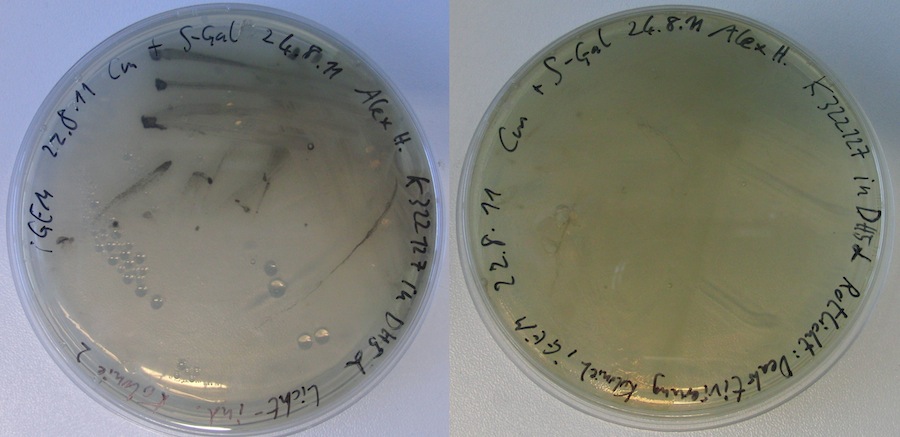
After over night incubation, both plates incubated in red light showed white colonies only. Of the two plates incubated under the lamp, one did not show any colonies. The other one showed black colonies. --> First evidence that part K322127 works as expected!!! To further quantify and characterise this: Miller assay? Other methods?
26-08-2011
People: Alex, Simon, Bea, Thorsten
Other Work
Part-request: Parts requested from the registry for backup if the ligation of the PCR product will fail again:
| Part | Team | Library | Plate | Well | Plasmid | Resistance |
|---|---|---|---|---|---|---|
| BBa_K322123 | iGEM10_Edindburgh | 2010 Submisions Samples 001 | Shipment: 00694 | 4D | pSB1C3 | C |
| BBa_K322124 | iGEM10_Edindburgh | 2010 Submisions Samples 001 | Shipment: 00694 | 4E | pSB1C3 | C |
And Gate plasmid: PCR-Product from Edinburgh
New media and plates: LB, LB for Agar plates (Kan and Amp?) and Luria medium were prepared. Plates were prepared.
Sequencing:
The reporter plasmid clone 5 was handed in for sequencing at GATC-Biotech. The primer used for sequencing is "Primer pSB1A2 und pSB1AK8 fwd".
Skype talk with iGEM Team Freiburg
Cloning
Miniprep & Glycero-Stock: The cultures inoculated yesterday on LB were frozen at -80 °C as glycero-stocks. The culture of K238013 (pSB1A2) in DH5alpha was prepped. The resulting DNA yielded a concentration of approximately 300 ng/ul.
Repetition of transformations:
The transformations from yesterday (ligation using ligation protocol) were repeated using DH5alpha cells (kindly provided by Andrea). Transformations were done for reporter plasmid (part 2b) colony 5 from DATUM, K238013 with lacZ, rbs with T7pol and T7prom with lacZ. After incubation for one hour, the cells were plated on LB Amp (K238013 with lacZ; rbs with T7pol) and on LB Kan (reporter plasmid; T7prom with lacZ) and incubated over night.
Ligation of new PCR product with pSB1A2
Material:
cut PCR (X,P) product from K322127 from PCR 25.8.2011 cut pSB1A2 (X,P) from Alex from 25.8.
procedure:
we purified PCR product using Zymo DNA Clean +Concentrator 25
ligation:
sample 1: 15µl PCR product 5µl vector sample 2: 10µl PCR product 10µl vector
Transformation of the ligation and R2 as transformation control:
the two samples from purifiying and the part R2( OmpR in pSB1A2) as a positive control were transformed via electroperforation in competent DH5alpha cells that were kindly supplied by Andrea Mückl.
Testing
Testing K322127
Material: inoculated cultures of DH5alpha with K322127 from 25.8 Procedure: 100 µl of the culture was spread on S-gal plates with Kan and amp in dark Incubation of the plates at room temperature (30°C) under light of diode with maximum of 630nm (turn-off diode) for 2 hours afterwards incubation at light at 37°C for 19 hours afterwards 4°C in the incubator: one culture with aluminium TUM logo put on top one culture in the box under light of turn-off diode (negative control) one culture on top of the box, light from turn-off diode from below, light from the incubator from top only on cut out TUM logo in aluminium foil
Results
26-08-2011
Transformations: The transformations from yesterday, including K238013 & lacZ, rbs & T7pol, T7prom & lacZ as well as PCR product & Vector, did not work at all. There were no colonies on the plates.
Sequencing: More data from the sequencing from 19-08-2011 arrived. The newly ligated K238013 & double terminator from 17-08-2011 does contain the double terminator. However, as witnessed before, the blue light promotor K238013, which was part of the plasmid prior to ligation, is missing. We are still awaiting the data from another sequencing from the same construct, but from another ligation.
Planning for next week: We need to prepare new electrocompetent DH5alpha cells. EVERYONE, who has free time to spare, please come to the lab and help. We might also be able to get new BL21 (DE3) cells from Prof. Buchner. If anyone wants to get them, contact Lars Mitschke at the Lehrstuhl of Prof. Buchner and talk to him about the request, which Alex asked about. BL21 (DE3) contain a T7polymerase, which can be induced by IPTG. This would be great use in order to test our reporter plasmid, i case the ligation worked (which we should know on Monday).
29-08-2011
Cloning
inoculation the following were inoculated: T7 Promotor with lacZ 2x K322127 in DH5alpha
Retransformation of reporter plasmid clone 5
the reporter plasmid clone 5 (118ng/µl) was diluted to 50 pg/µl and to 20 ng/µl
1 µl of each dilution was transformed into DH5alpha
Transformation of RBS
0,42 µl of RBS (47,5 ng/µl) were transformed into DH5alpha
Results
26-08-2011
transformations
were only successful for R2, T7 Promotor with lacZ, reporter plasmid colony 5
testing of K322127
only very small colonies on the plate with no coloring plate that was illuminated with turn-off light and light from the incubator dried out. no black colonies could be detected. Incubation of plate with TUM pattern was continued. Plate was irradated with a light bulb.
sequencing of reporter plasmid colony 5

Bases in squared brackets are optional and belonging to another sequence. Due tu overlapping base shifts exact assignment of bases is difficult
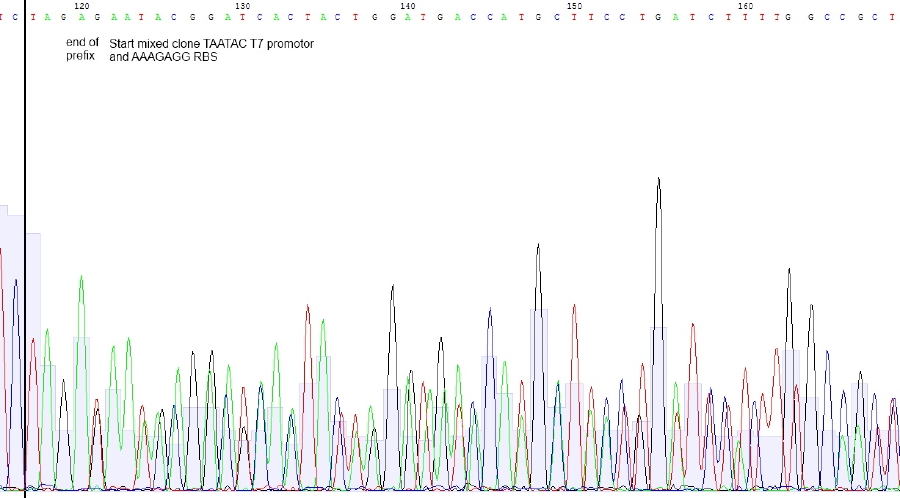
Sequencing reveals mixed clones. Correct Sequence with T7 promotor - RBS – lacZ is probably present beneath construct with T7 promotor lacking. Preparation of RBS-LacZ vector was contaminated with uncut or religated vector. Since plasmid length was ok, construct should be present.
next steps:
Retransformation with 50 pg and 20 ng MiniPrep DNA will be performed with subsequent sequencing of 4 clones. In parallel new construct from Alex will be sequenced.
30-08-2011
People: Thorsten, Bea
Results
29.8.11
Re-Transformation of part 2b clone 5 with 20 ng vector was succesful. 50 pg vector didnt yield any colonies. RBS was tranformed succesfully. 5 ml LB o/n cultures with apropriate antibiotica were inoculated and incubated at 37° C shaking. Mini Prep and sequencing will be performed at 31.8.11
Cloning
Electrocompetent cells
- new electrocompetent cells (DH5alpha) were made by the following protocol:
- a 20 ml Luria medium overnight culture was inoculated with electrocompetent DH5alpha (kindly provided Andrea Mückl) yesterday
- 350 ml Luria medium were inoculated with 15 ml of the overnight culture and incubated at 37°C until a OD(600) of 0,7 was reached
- the medium was distributed on falcons and the cells were centrifuged at 4500 rpm for 20 min at 4°C
- after discarding the supernatant 350 ml of sterile ice-cold 10% glycerol was added and the pellets were resuspended using a pipet
- the resuspension was centrifuged at 4500 rpm at 4°C for 11 minutes
- after discarding the supernatant the cells were resuspended again in 350 ml of ice-cold 10% glycerol ( cells kept chilled all the time)
- and again centrifuged at 4500 rpm at 4°C for 11 minutes
- the supernatant was discarded and the cells were resuspended in the remaining supernatant
- the cells were dispensed in 40 µl aliquots and stored at -80°C for further use
Planning
In order to get ligations working, purification steps will be performed using Promegas Wizard PCR and Gel clean up Kit. We are going to ligate PCR product into pSB1C3 and upstream of SupD-tRNA. In parallel, T7ptag will be ligated into AND-Gate Plasmid, which will be pSB1C3. [First step of assembly]
Then, synthesis product, which contains parts from SupDtRNA to RBS J44001, will be ligated into succesful AND-Gate Plasmid-T7ptag. The last step will be ligation of PCR-product into this construct.
Excel sheets can be found in DropBox folder "protocols".
planned parts
PCR product ligated into pSB1C3 will be a new part "red light sensor without lacZ".
PCR product ligated with SupD will be a new part as well.
Synthesis product is another maybe not so useful part which will be send into the registry.
Reporter plasmid will be another Part. This one was called "part 2B" in recent cloning steps and needs further validation (see 29.8. retransformation and sequencing)
Restriction digest
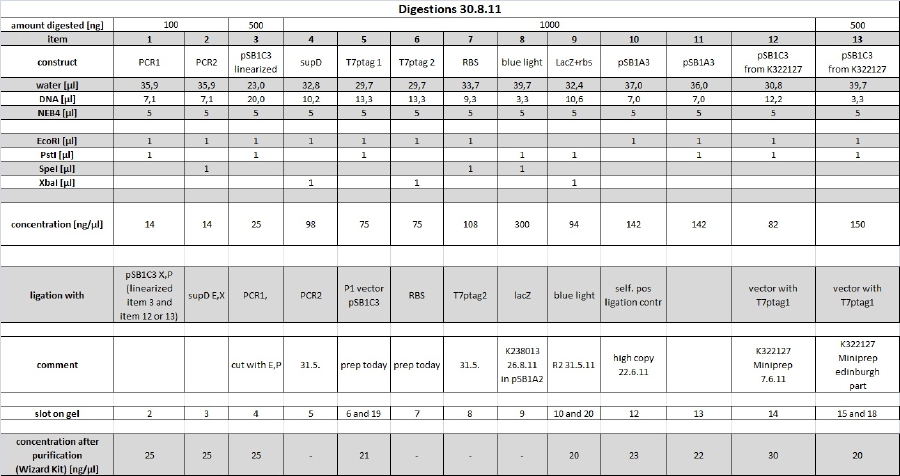
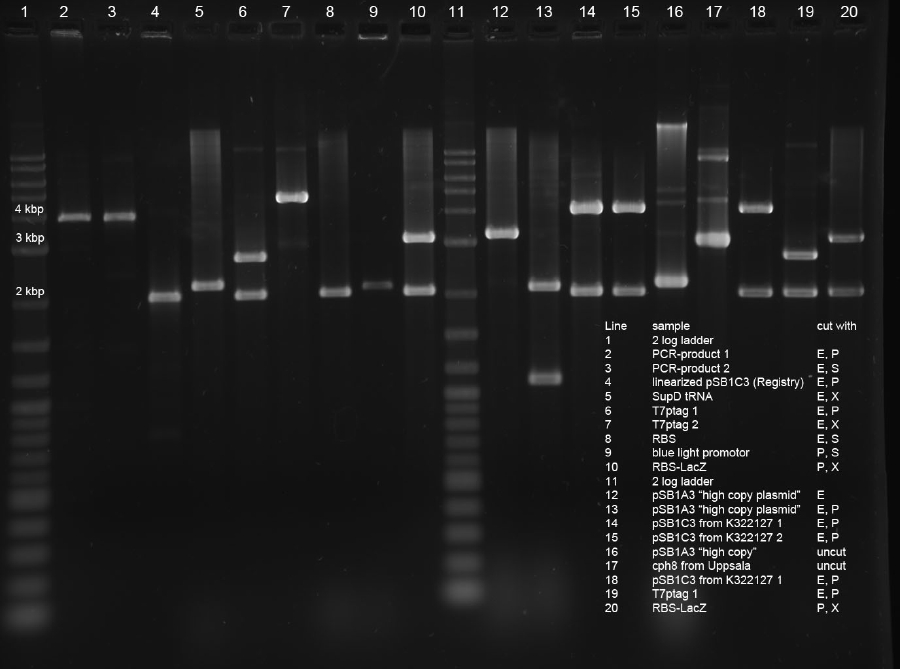
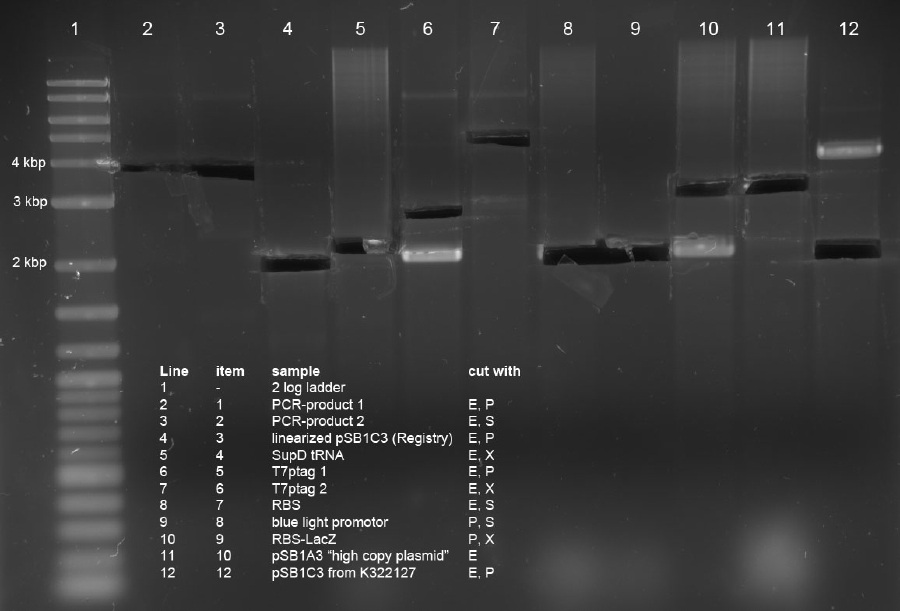
All digestions were complete and running on the expected lengths.
Purification of restricion digests from gel with Wizard Kit
the following items were purified using the Wizard Kit according to manufacturer's instructions and the concentration was determined by nanodrop:
1: 24,5 ng/µl
2: 24,5ng/µl
3: 24,5 ng/µl
5: 20,5 ng/µl
9: 20,0 ng/µl
10: 23 ng/µl
11: 21,5ng/µl
12: 29,5 ng/µl
13: 20,0 ng/µl
for more details see restriction digest table
Inoculation
the following overnight cultures were inoculated:
RBS reporter plasmid clone 1-5 K322127 from plate from 24.8.2011
31-08-2011
Results
30.8.11
Electrocompetent cells show good transformation efficiency. 100µl 10µl and 1µl of test transformation of 30 ng BBa_K322127 were plated on LB/cam plates. 1 µl yielded ca 400 colonies which refers to a tranformationefficiency of 1.3*10^7 cfu/µg BBa_K322127 in pSB1C3. 1*10^8 cfu/µg pUC19 is expected, so 1.3*10^7 cfu/µg vector with insert seems to be ok.
cph8 from Uppsala was succesfully transformed into DH5alpha. Cells have been 1:3 diluted and 20 ng plasmid was used. Output was ca 10^6 - 10^7 cfu/µg. Plate will be stored at 4°C til inoculation of o/n culture with subsequent minipreparation.
Cloning
Ligation
Minipreparation of DNA
The following concentrations were gained through minipreparation:
RBS : 86,5 ng/µl
reporter plasmid clone 1: 135 ng/µl
reporter plasmid clone 2: 234 ng/µl
reporter plasmid clone 3: 102 ng/µl
reporter plasmid clone 4: 119 ng/µl
reporter plasmid clone 5: 96 ng/µl
The reporter plasmid clone 1-5 were sent to be sequenced at GATC.
Testing
the overnight culture of K322127 was used to inoculate a 100 ml culture in LB which was incubated for 5 hours at 37°C
at an OD of 1.6 50 ml of the cells were centrifuged at 4500 rpm the supernatant discarded
the cells were resuspended in remaining 1 ml of LB
100 ml of LB media with s-gal was prepared and autoclaved according to protocol see Methods
the resuspended cells were added to the medium when it reached 40°C
the media was given in thee plates
two plates were incubated at 37°C under turn-off light (diode max. 630 nm)
the TUM cookie cutters were inserted in one plate that was then incubated under the light of a lamp
Other Work
new chloramphenicol plates were made
01-09-2011
Cloning
gel purification of 30.8.11 digest
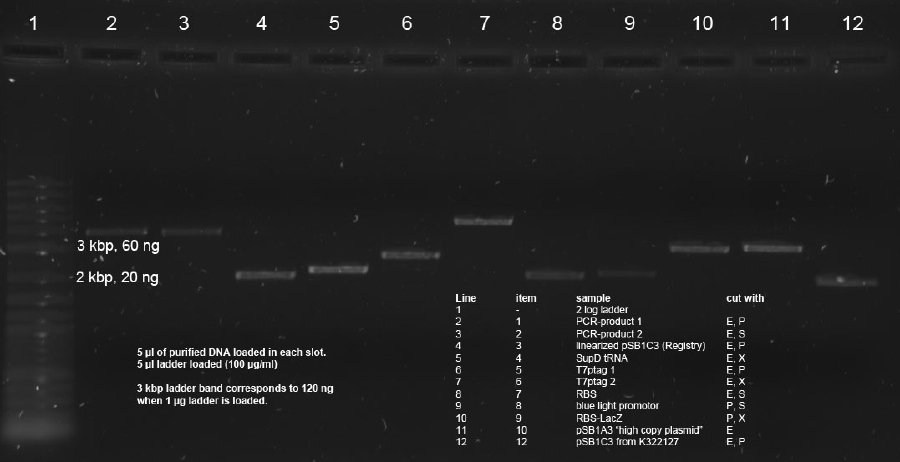
20 µl of remaining digestions (made on 30.8.11) were loaded on 1 % agarose gel with 1xTAE. Items 1,2,3,4,5,6,7,8,9,10,12 were used. This samples were cut using 350nm UV illumination as short as possible. Cut gel slices were subsequently purified using Promegas Wizard Kit according to manufacturers instructions.
Determination of concentration with Nanodrop resultet in not reliable data. Therefore quantitative 0.7 % agarose gel was run with 5 µl of each sample. 5 µl 2 log ladder was used (100 µg/ml). PCR Band (3.8 kbp) in line 2 was estimated to 75 ng. This gives aproximately 15 ng/µl. Intensities of 2 kbp band in line 4 as well as lines 5, 6, 7, 10 and 11 correspond to approx. 150 ng amount (--> 30 ng/µl).
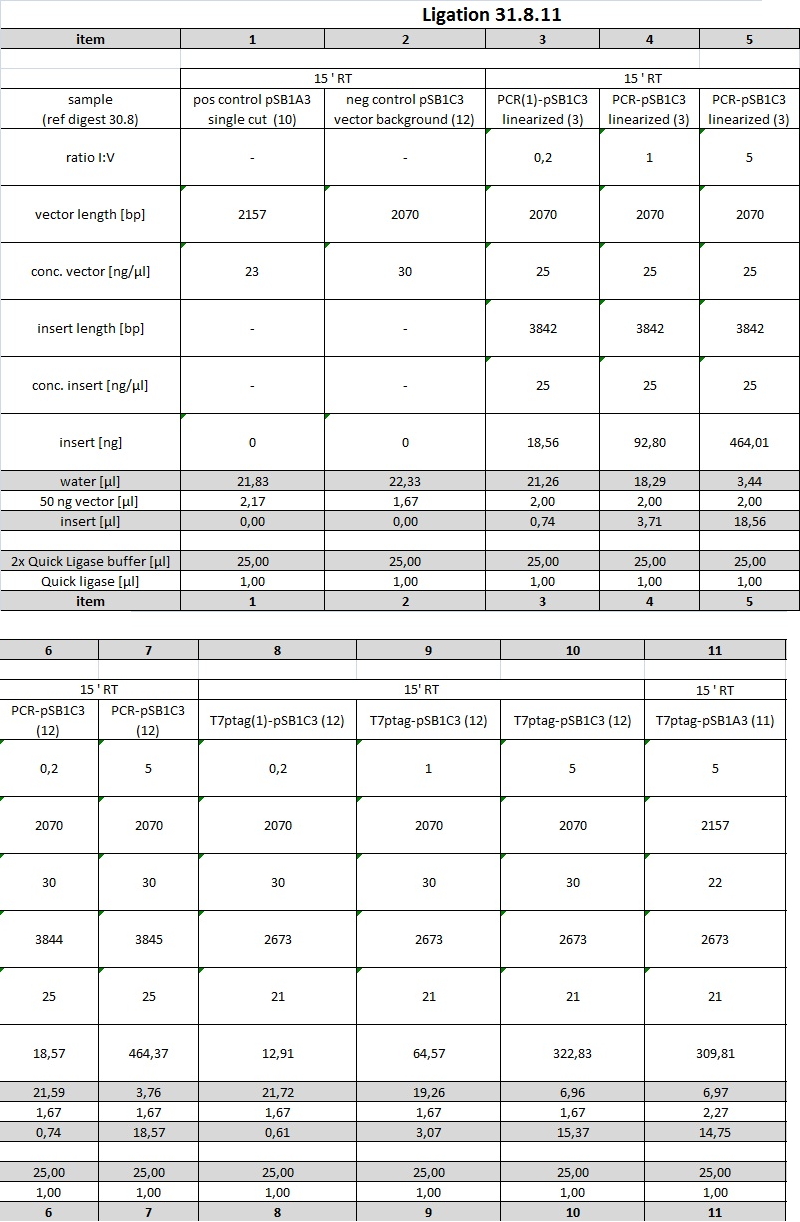
Due to fly-by-night intensity of ladder those concentrations will be determined again on 02.09.11 for further ligations. Ligations today will be done with the following concentrations

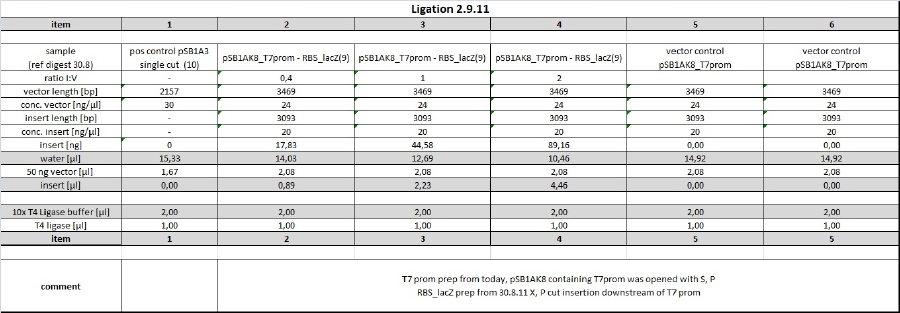
o/n ligation with T4 Ligase
P, S opened Vector containing blue light promotor was ligated with RBS-LacZ insert. Due to huge insert lengths, insert:vector ratios were choosen as can be seen in ligation table.
Ligation was conducted at 16° C in thermocycler o/n using 20 µl sample volume and 1 µl T4 DNA ligase. Examplary calculation of I/V ratio for 6x molar excess of insert (source: http://openwetware.org/wiki/DNA_Ligation):
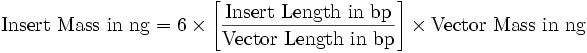
Testing
inoculation of two cultures with 5 µl of the glycerol stock of K322127 in DH5alpha
the cultures were incubated at turn-off light (diode max. at 630 nm) at 37°C
new transformation of K322127 (150ng/µl) 0,5 µl were used
Testing
Preparing lacZ-ONPG or also called Miller Assay
Other Work
Part design in the registry
edit the general information of the part : http://partsregistry.org/wiki/index.php?title=Part:BBa_K568000&action=edit
others
the primer for the sequencing of reporter plasmid clone 1-5 from yesterday is sent to GATC
Lars Mitschke kindly provided the E. coli strain BL21 (DE3), this strain contains a T7 Polymerase which can be induced with IPTG.
Results
31-08-2011
transformations
no colonies on the plates for all ligations
testing of K322127
the plate that was incubated under light showed no coloring but the plate became intransparent which indicates that the bacteria grew
plates incubated under turn-off light (diode max. 630 nm) became intransparent as well; this plates were used for further testing ( see testing of K322127 )
02-09-2011
Cloning
Digest of T7 promoter
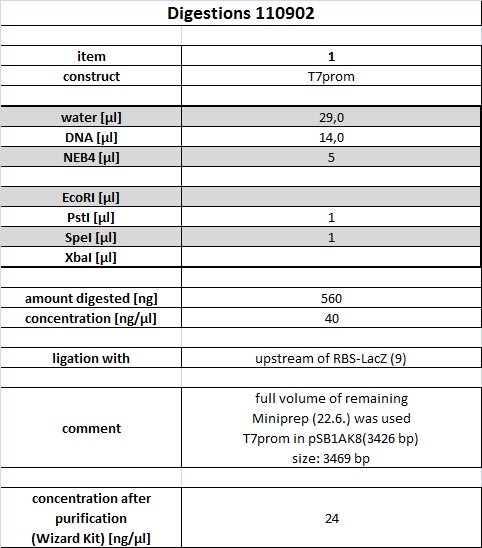
Full volume of digest was loaded onto gel (50 µl digest + 10 µl 6 x Gel loading buffer)
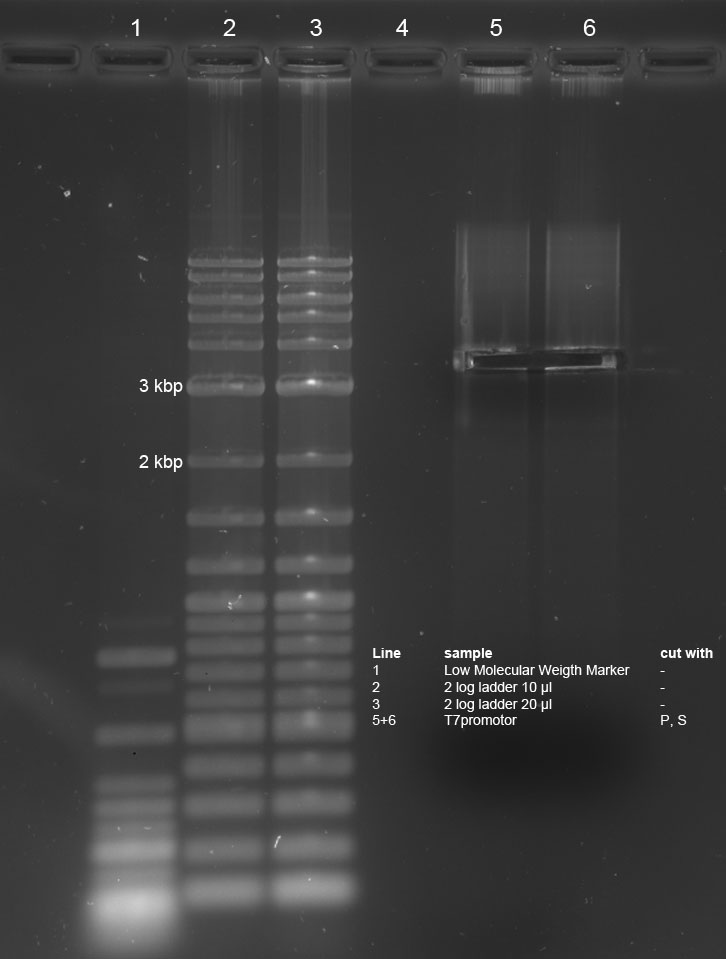
Band with correct size of 3,5 kbp was cut using 365 nm illumination. Illumination time was kept as short as possible, picture was taken after cutting gel slice.
Gel purification of T7 promotor
Band was purified using Promega's Wizard Gel purification Kit according to manufacturer's instructions. Eluted DNA was subsequently quantified with an analytical 0.7 % agarose gel (120 V for 45 min)
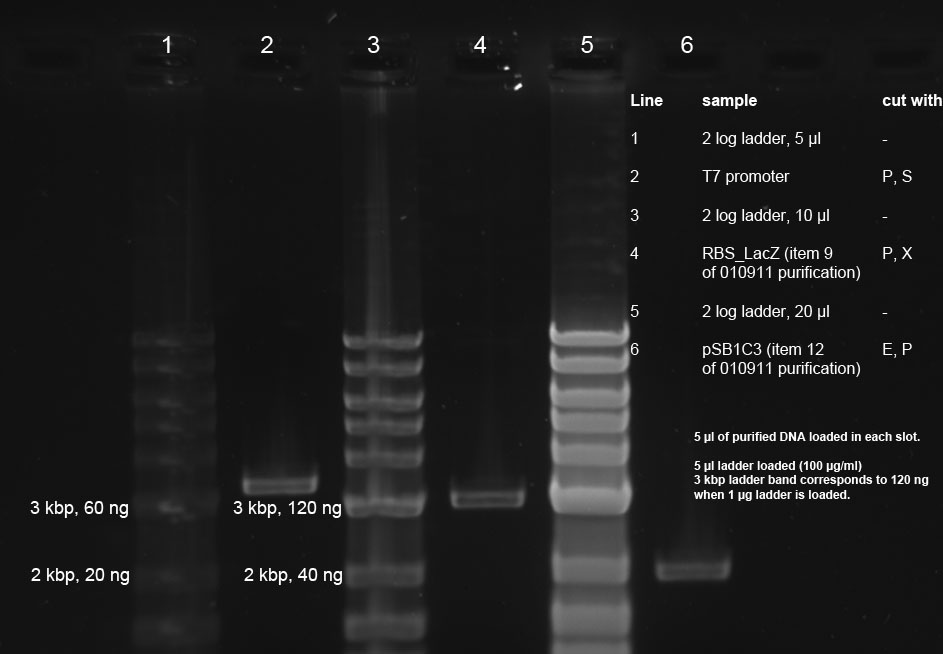
Band in line 2 corresponds to 120 ng band of marker. Therefore concentration of purified T7 promoter is approx. 24 ng/µl. RBS_lacZ band is approx. 20 ng/µl and pSB1C3 band approx. 16 ng/µl
Ligation of Reporter Plasmid
digested T7 promoter was ligated with RBS_lacZ from 1.9.11 purification.
Ligation was conducted o/n at 16°C.
Other Work
Agar Plates
500 ml of media for ampcillin plates
250 ml of media for s-gal plates were prepared
Cloning
Transformation
the ligations from 01-09-2011 (1-7) were transformed into DH5alpha as a transformation control 0,5 of R2 (233ng/) was transformed into DH5alpha
Testing
preparing ONPG assay
ONPG solution (4mg/ml)
1 M Na2CO3 were prepared
Overnight Cultures
new overnight cultures of the new transformation of K322127 (see 01-09-2011) were inoculated
Results
01-09-2011
Testing K322127
no coloring on plates
03-09-2011
People: Bea, Thorsten, Susan, Wolfgang
Other work
Cloramphenicol stock was produced: 30 mg/ml final concentration in 100% ethanol. 153 mg + 5,1 ml 100% EtOH. 500 µl aliquotes were stored at -20°C.
Cloning
Precipitation of Ligations and Transformation
Todays Transformation were done after purifing ligation mix with Glycogen/Ethanol precipitation in order to increase transformed amount and purity of ligated vectors.
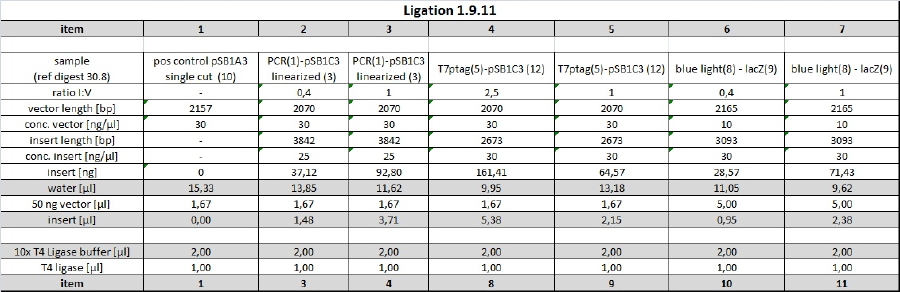
All Ligations of 02-09-11 and following items of 01-09-11 ligation were used:
- 1 (pos control pSB1A3single cut (10))
- 2 (PCR(1)-pSB1C3 linearized (3))
- 4 (T7ptag(1)-pSB1C3 (12))
- 6 (blue light(8) - lacZ(9))
Glycogen/ethanol precipitation:
- -20 µl of the ligation mixed with 2 µl NaAc (3M), 0.5 µl glycogen and 22.5 µl isopropanol
- -incubation step for 1 h @ -20 °C
- -15 mins @ 10.000 rpms
- -withdraw supernatant
- -wash pelett with 70 % EtOH ~ 10 µl
- -dry pelett and resuspense in 10 µl water
Transformation via electroporation was done with the whole cleaned 10 µl in 40 µl DH5alpha cells.
Ligations of PCR product into pSB1C3 and pSB1A3
Following ligations were examined using an excess of vector and 1 µl PCR-product (corresponds to approx 14 ng/µl).
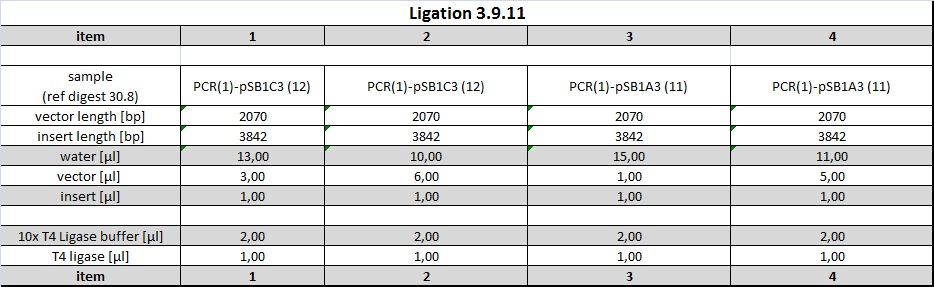
Items 1+2 were incubated at room temperature and items 3+4 were incubated at 37° C for 1h each. 5 µl of ligations were subsequently electroporated into 40 µl undiluted DH5alpha cells.
Results
02-09-2011
Testing
no coloring on plates
Cloning
Single cut vector control(1) showed approx. 50 colonies. ALl ligations plated on Cam plates (2-5) showed no colonies.
Blue light - RBS_lacZ ligations (6+7) showed 5 colonies overall. Clones were inoculated into 5 ml LB/Amp and incubated at 37°C o/n.
04-09-2011
People: Bea, Thorsten, Flo
Results
03-09-2011
Transformation of purified ligations (02-09-2011)
positive control showed several thousand red colonies. All ligations showed bacterial lawn, including vector control.
Vector prep contains uncut or single cut vector. This result is consistent with sequencing results, which indicate mixed clones. Next steps: Preperation of 46 bp T7 promoter insert with 3 % preparative agarose gel and ligation upstream of RBS_lacZ into opened RBS_lacZ vector. In parallel vector prep of T7 promoter will be repeated on 1% agarose gel with a long gel and run. Uncut and single cut vector will be loaded on gel aswell.
Transformation of 03-09-2011 ligations
showed following output:
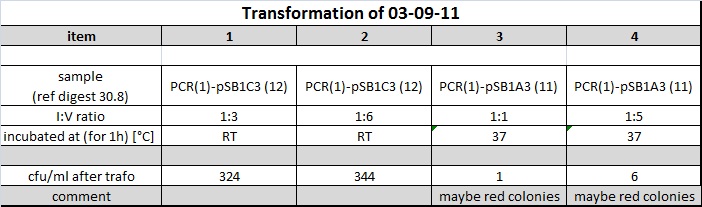
pSB1A3 colonies were possibly red. --> incubated again o/n
http://partsregistry.org/Part:BBa_J04450 --> red color indicates religated or uncut vector clone
4 clones of each successful transformation were picked and inoculated into 5 ml LB medium containing the appropriate antibiotica.
Conclusion:
Further incubations should be done at RT for 1h or o/n at 16°C with subsequent gylkogen/ethanol purification and transformation of whole 10 µl resuspended pellet into 40 µl DH5alpha
Cloning
Reporter Plasmid
As recent transformation of reporter plasmid yielded high vector background new preps will be done according to following scheme.
digest
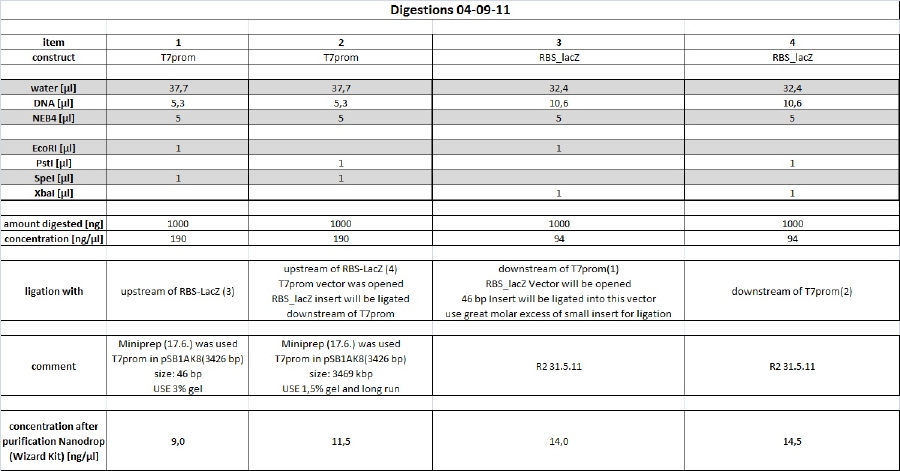
long 1% preperative agarose gel and 3% preperative agarose gel have been produced and stored in a plastic bag with some 1x TAE buffer at 4°C o/n. Purification will be done tomorrow.
Minipreps of picked clones of Bluelight_lacZ 01-09-11 ligation (items 6+7)
[Flo]
Miniprep of 5 o/n cultures [names below protocol] (Metabion Kit)
All centrifugation steps were performed at 13.000 rpm and RT
1. Pellet 2 x 1,5 ml of bacterial suspension in one 2 ml-Eppi, centrifuge for 1 min each
2. Add 250 ul Bact.resuspension buffer, vortex
3. Add 250 ul Cell lysis buffer, invert 5x, incub. approx. 4 min
4. Add 350 ul DNA binding buffer, invert 5x, centrifuge for 10 min
5. Transfer clear lysate on column, centrifuge for 1 min, discard supernatant
6. Add 600 ul Column Wash Buffer, centrifuge for 1 min
7. Transfer column to new 1,5 ml-Eppi
8. Add 50 ul nuclease-free water (from Promega Wizard Kit), incub. approx. 2 min, centrifuge for 1 min
9. Store at -20°C
names:
-Blue Prom + lacZ #1 (= former "blue lacZ Trafo 7, Klon 2")
-Blue Prom + lacZ #2 (= former "blue lacZ Trafo 7, Klon 1")
-Blue Prom + lacZ #3 (= former "blue lacZ Trafo 6, Klon 1")
-Cph8 in DH5a
-K322127
Preparing glycerol stocks from cultures used above
Prepare 50% glycerol/LB solution by mixing 2 ml 100% glycerol and 2 ml LB
Add 300 ul of 50% glycerol/LB solution to 700 ul bacterial suspension to obtain an end concentration of 15% glycerol
Store at -80°C
Restriction digest of 5 Minipreps from today
Each plasmid has been cut with EcoRI HF AND EcoRI HF + PstI HF (= 2 digests per plasmid) as follows:
-5 ul Plasmid DNA
-2 ul 10x NEB buffer 4
-0.5 ul EcoRI HF
-(0.5 ul PstI HF)
-12 (11.5) ul ddH2O
end volume is 20 ul
mix, spin down, incub. at 37°C for 1 hour, inactivate at 80°C for 20 min
Add 4 ul 6x loading dye
Store at -20°C until tomorrow
--> quantification on gel 05-09-11
Testing
ONPG-Test - Miller Assay
The assay was conducted following the protocol:
3 samples of K322127 in DH5alpha were assayed: 1 sample was incubated for 25 hours in the dark at room temperature 1 sample was incubated for 25 hours in the light at room temperature 1 sample was incubated for 25 hours in the turn-off light at room temperature
- - inoculation of 4 x 2ml LB with 10 μl of overnight cultures of K322127 (see 02-09-2011)
- - light induction when the cultures reach OD of around
- - control with only LB
start of Assay:
- - measurement of Abs(600nm)
- - new eppi with 1 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood)
- - 100 μl of the samples are taken and transferred to the Zbuffer
- - mix the solution by vortexing for 10 s
- - let the chloroform settle to the ground and take 700 μl of supernatant for measurement
- - initiation of assay with 140μl of ONPG (4mg/ml) NOTE START TIME
- (in the paper they use 100 μl supernatant plus 20 μl of ( 4mg/ml but we need more for measurement in cuvette)
- -incubation at room temperature until yellow color develops
- -stop reaction with 350 μl of 1 M Na2CO3 NOTE STOP TIME (we upscaled the paper uses 50 μl)
- - measurement of Abs (420nm) and Abs(550nm)
Abs 420 nm should be less than 1 and more than 0,5
Result
incubation for 146 min because no visible yellow color developed;
not completely clear how to calculate the miller units:
here miller units are calculated as following:
units= 1000* (Abs 420-1,75 Abs550) /(time(min)*Abs 600/(11,6*1,7)))
Results
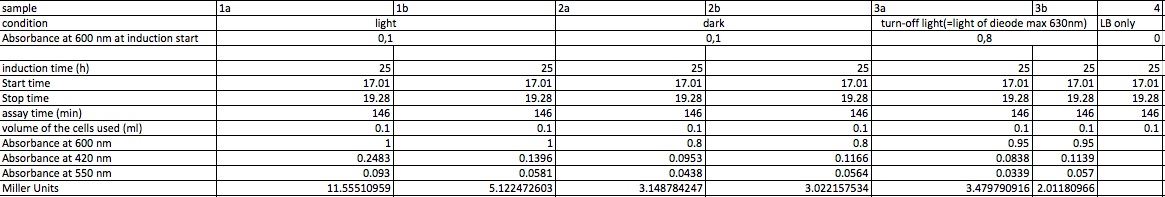{kind=link}
Other Work
S-Gal plates
Flo did Minipreps of cph8 from Uppsala and K322127. Nanodrop yielded no reliable data. --> quantification on gel
05-09-2011
People: Bea, Thorsten, Anna
Testing
Miller Assay
5 newly inoculated cultures were assayed:
induction start means: incubation at room temperature of: K322127 in DH5alpha in the light of lamp K322127 in DH5alpha in the dark K322127 in DH5alpha in the turn-off light K322127 in BL21in the light cpH8 in DH5 alpha in the light
a new protocol was used for this assay:
start of Assay:
- measurement of Abs(600nm)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood)
- 0,5 ml of the samples are taken and transferred to the Zbuffer
- mix the solution by vortexing for 10 s ( all samples equal vortexing time)
- initiation of assay with 200μl of ONPG (4mg/ml) NOTE START TIME
-incubation at room temperature for see table
-stop reaction with 500 μl of 1 M Na2CO3 NOTE STOP TIME
- measurement of Abs (420nm) and Abs(550nm)
Results
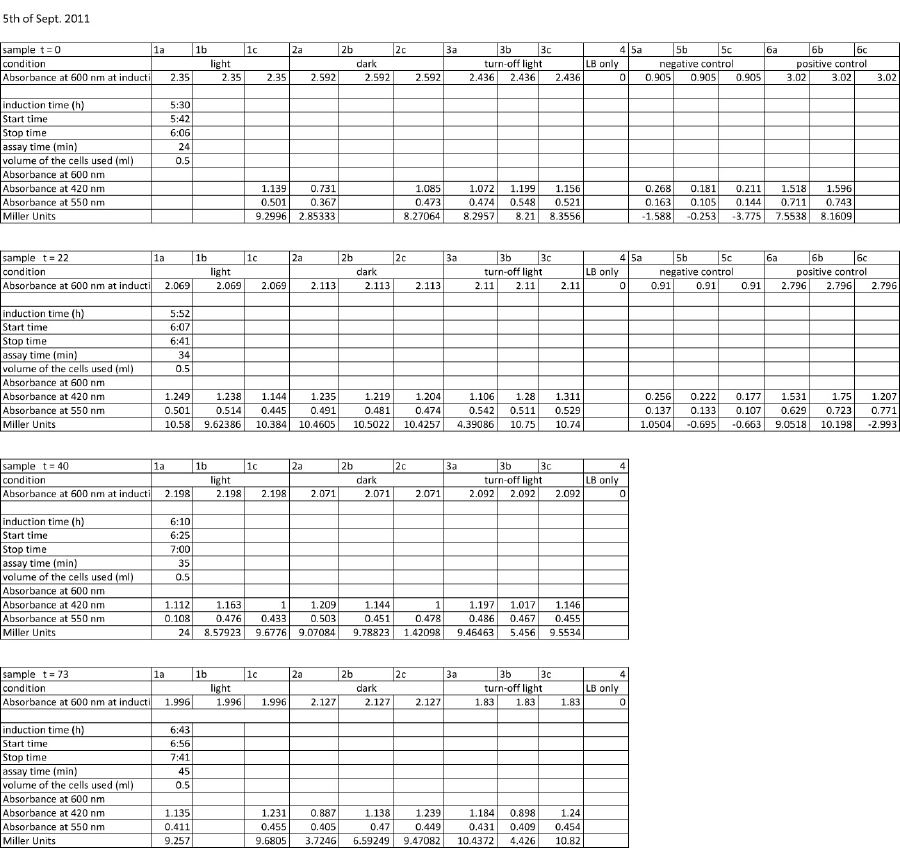
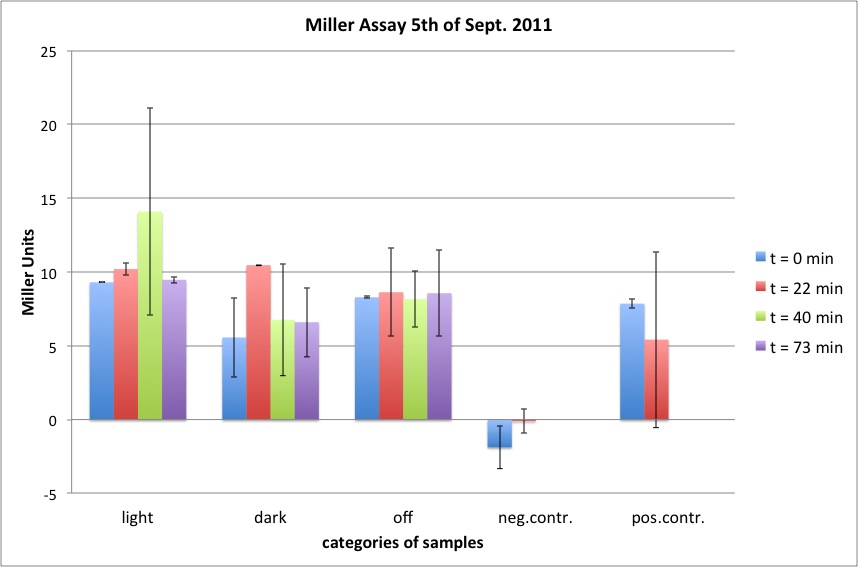
Cloning
Analytical gel of Bluelight_lacZ, cph8 from Uppsala and K322127 testdigest
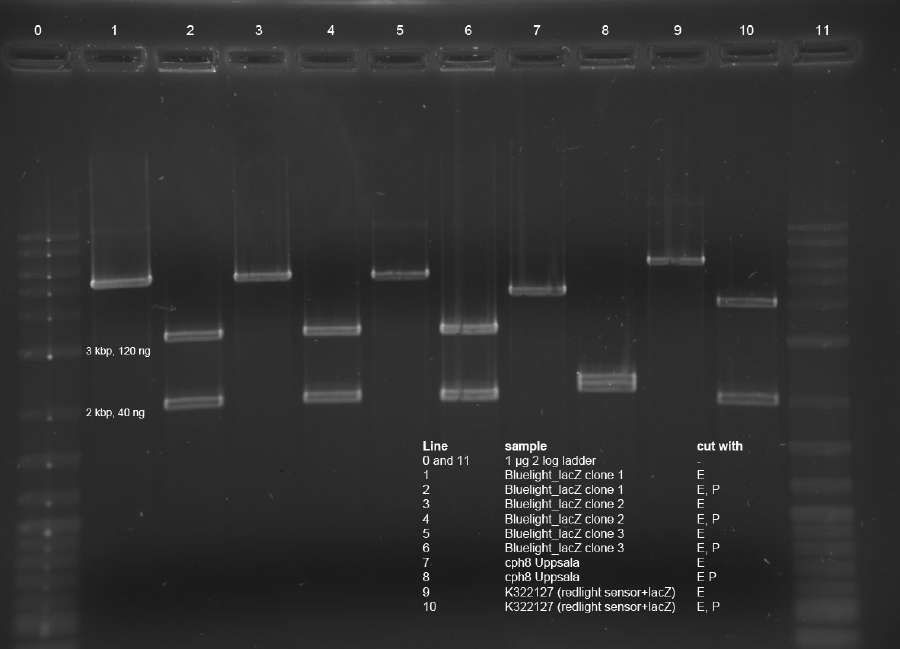
Size of bands look fine. Expected: 3093bp(lacZ)+86bp(Bluelight)= 3179 bp
--> Send clone 2 for sequencing or load RBS_lacZ digest along with ligated construct on gel and check for 86 bp difference.
cph8(2,2kbp) and K322127(4kbp) are running on their correct sizes.
Miniprep and testdigest of picked colonies on 04-09-2011
Miniprep of yesterdays picked clones were done. Concentration was determined using Nanodrop.
T7 Pol in pSB1C3 (ligation 4 from 1.9.)samples:
clone 1 : 58,5 ng/µl (sample 1)
clone 2 : 60,5 ng/µl (sample 2)
clone 3 : 55,5 ng/µl (sample 10)
clone 4 : 56,5 ng/µl (sample 13)
PCR in linearized pSB1C3:
clone 1 : 44,0 ng/µl (sample 4)
clone 2 : 82,0 ng/µl (sample 7)
clone 3 : 77,5 ng/µl (sample 5)
clone 4 : 70,0 ng/µl (sample 14)
blue light promotor+ lacZ (ligation 6 from 1.9):
clone 1 : 79,0 ng/µl (sample 12)
clone 2 : 67,0 ng/µl (sample 9)
clone 3 : 55,5 ng/µl (sample 21)
clone 4 : 65,0 ng/µl (sample 19)
red light sensor in pSB1C3 ratio 1:3
clone 1 : 58,0 ng/µl (sample 11)
clone 2 : 52,5 ng/µl (sample 15)
clone 3 : 59,5 ng/µl (sample 8)
red light sensor in pSB1C3 ratio 1:6
clone 1 : 50,5 ng/µl (sample 18)
clone 2 : 58,5 ng/µl (sample 3)
clone 3 : 61,5 ng/µl (sample 6)
red light sensor in pSB1A3 (ligation 1:5 (from 1.9.) clone 1 : 52,5 ng/µl (sample 20)(red colony) clone 2 : 51,0 ng/µl (sample 16)
red light sensor in pSB1A3 (ligation 1:1 (from 3.9) (red colony): clone 1 : 40,5 ng/µl (sample 17)
All samples were digested except 9,21,19:
5 µl of Mini-DNA was cut using 0,5 µl E and 0,5 µl P in 2 µl NEB4 and 20 µl total volume. Digest was loaded onto 1 % agarose gel.
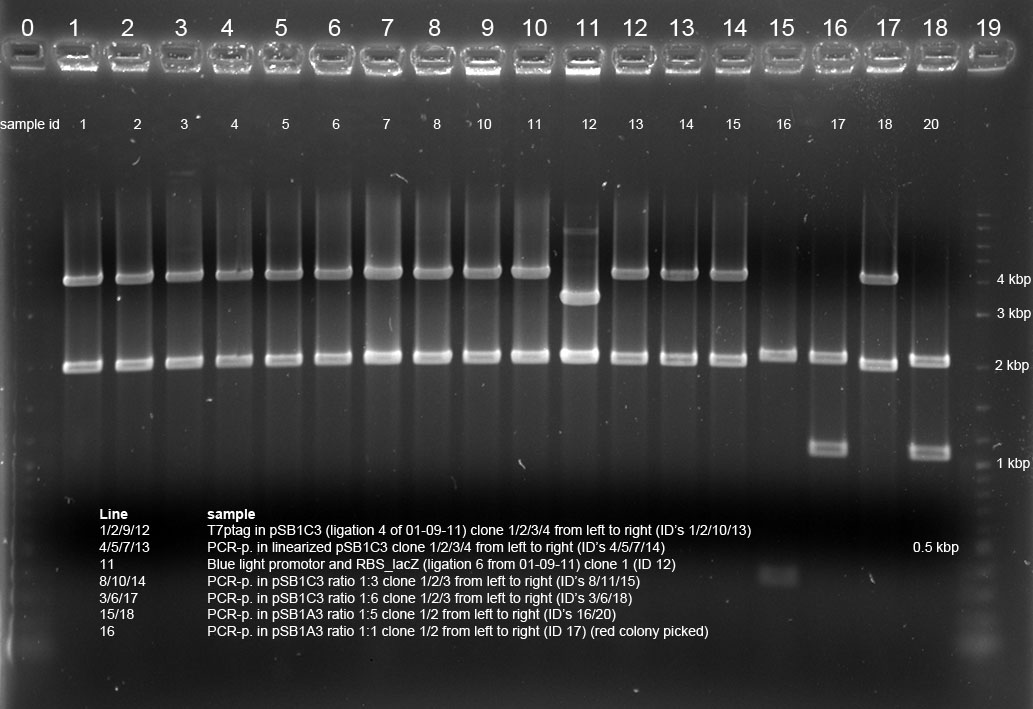
Red light sensor constructs look fine (3,8kbp insert and 2 kbp vector). T7ptag constructs are wrong and will be repeated (T7ptag expected at 2,6 kbp). Blue light sensor looks fine (3kbp). Red light sensor cloning into pSB1A3 didnt yield any insertion products, mainly slightly red colonies were visible --> 1kbp insert is RFP cassette. One not-red clone could be picked (sample 15), which contains a 400 bp insert of unkown origin.
--> Sample 7 was choosen to be sequenced as it yielded most amount of DNA.
Reporter Plasmid: T7 promotor prep and new RBS_LacZ prep
3 % agarose gel from yesterday was used at 100V for 1,5h
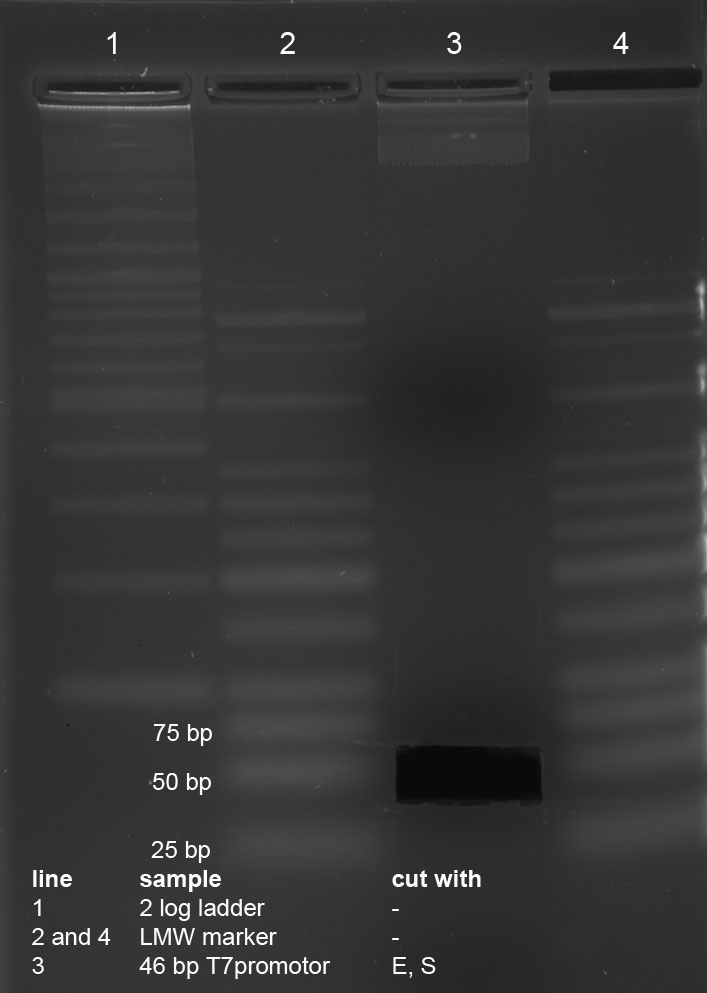
No band could be detected. Nevertheless, gel slice was cut and will be purified.
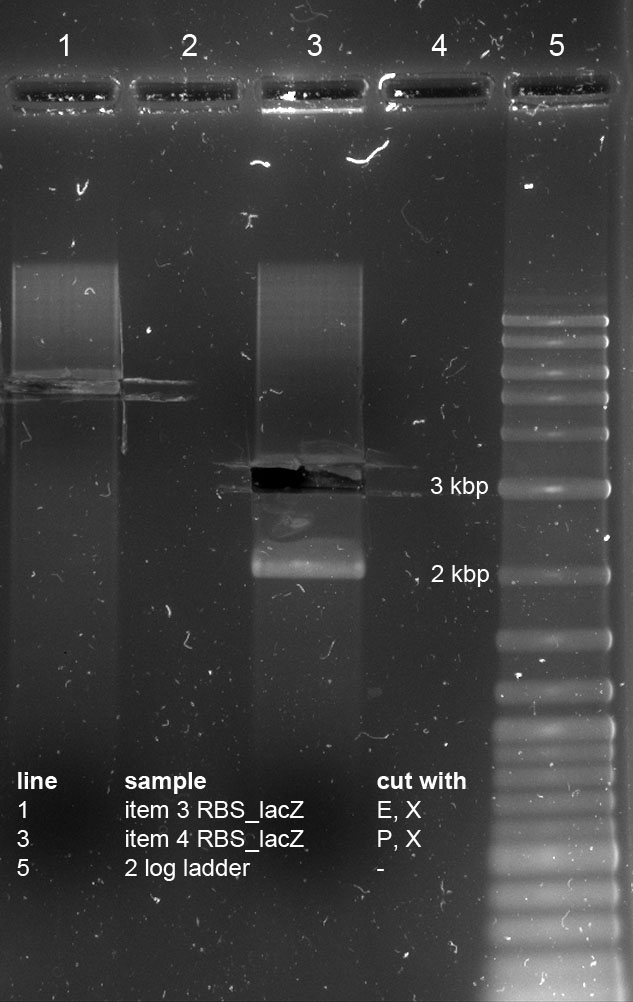
Bands were cut and purified using Wizard Kit according to manufacturer's instructions.
Concentrations was determined using Nanodrop:
T7prom E, S: 9 ng/µl
T7prom P, S: 11.5 ng/µl
RBS_lacZ E, X: 14 ng/µl
RBS_lacZ E, X: 14.5 ng/µl
06-09-2011
People: Thorsten, Anna, Katharina
Testing
Miller Assay
5 newly inoculated cultures were assayed: difference to day before: grow in the dark (only DH5alpha)
incubation at room temperature of:
K322127 in DH5alpha in the light of lamp
K322127 in DH5alpha in the dark
K322127 in DH5alpha in the turn-off light
K322127 in BL21 in the light
cpH8 in DH5 alpha in the light
Problem 1: OD of bacterial cultures was very high, should have been around 0,7.
Problem 2: too long incubation of color reaction yielded too high ODs, samples couldn't be diluted as more blank sample (as diluent) was missing.
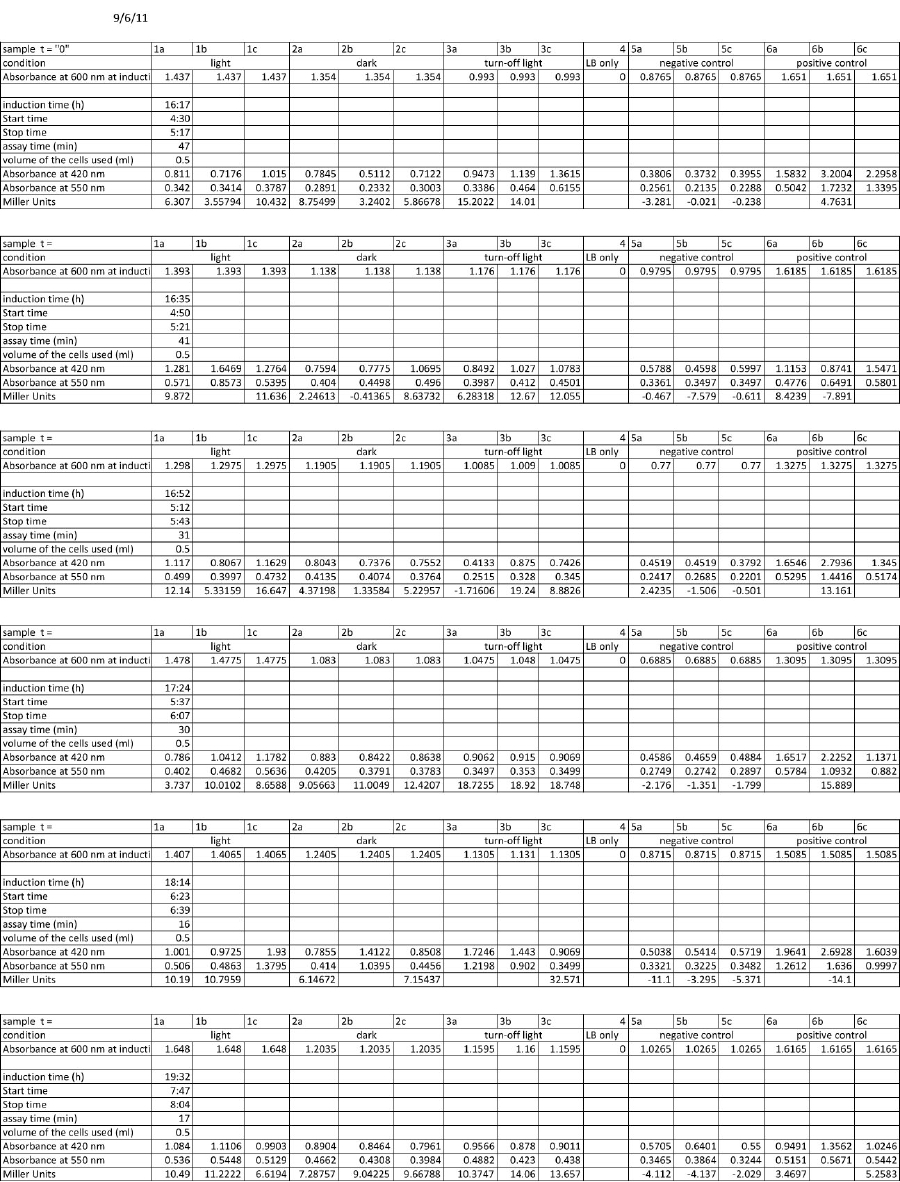
Results
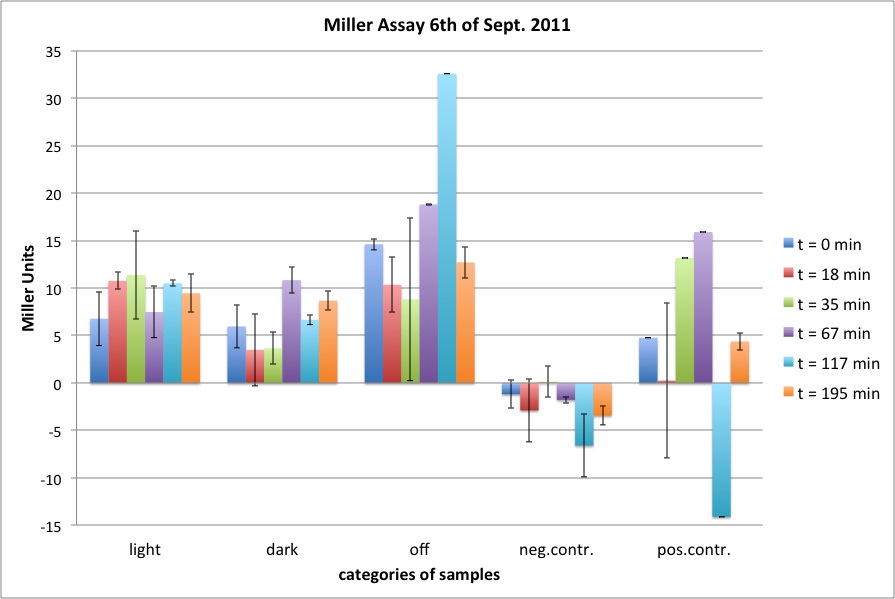
Cloning
Reporter Plasmid
Purification of 05-09-11 gel runs
Frozen gel slices were purified using Promega's Wizard Kit according to manufacturers instructions.
Subsequent determination of concentration via Nanodrop:
1: T7prom 46 bp cut: 9 ng/µl
2: T7prom vector cut: 11.5 ng/µl
3: RBS_lacZ vector cut: 14.0 ng/µl
4: RBS_lacZ insert cut: 14.5 ng/µl
Ligation of purified samples: Reporter Plasmid and T7ptag into shipping vector pSB1C3
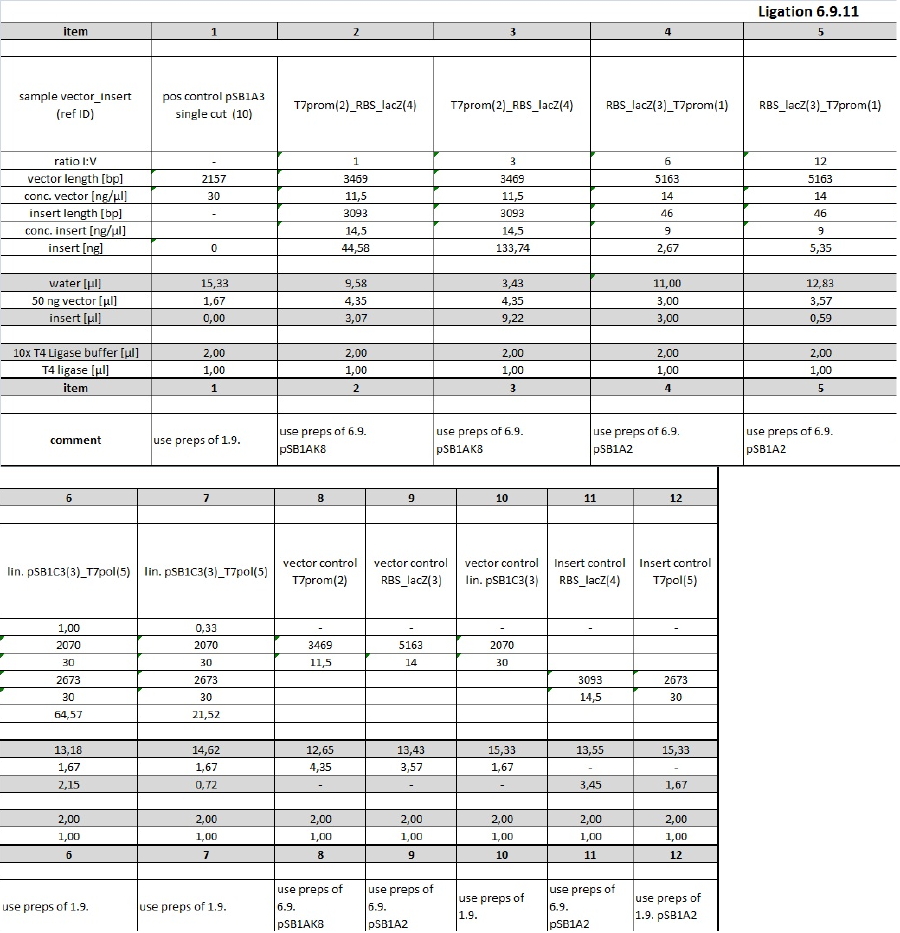
Ligation mix was incubated for 1h at RT and and then at 16°C o/n.
Other Work
Sequencing of clones
Primer overview: http://partsregistry.org/Primers/Catalog
For most biobrick plasmids the standard sequencing primers VF2 (verification forward) and VR (verification reverse) will fit. They will be synthesized at GATC and stored for 1 year.
10 µl of Following clones were send for sequencing 300-100 ng/µl with VF2 primer (tgccacctgacgtctaagaa):
Bluelight_lacZ in pSB1A2 110904_clone2 miniprep conc.: 49.5 ng/µl
red light in pSB1C3 110905_sample 7 conc.: 82 ng/µl diluted 1:1 with ddH2O
07-09-2011
People: Thorsten, Anna, Katharina
Cloning
Reporter Plasmid and T7ptag into pSB1C3
Glycogen/ethanol precipitation and transformation
- DNA precipitation with glycogen:of the ligation samples from 6-9-11
1. add 2 µl 3 M sodium acetate, 0.5 µl glycogen and 22.5 µl isopropanol to 20 µl ligation product
2. incubate the mixture at -20°C for one hour
3. centrifuge the mixture for 15 minutes at 18,000 rpm, 4°C
4. discard the supernatant
5. add 50 µl 70 % ethanol
6. centrifuge the mixture for another 10 minutes at 18,000 rpm, 4°C
7. remove supernatant with a pipet CAREFULLY. If pellet moves, centrifuge again.
8. air-dry the pellet until the ethanol is completely evaporated. Do not over-dry pellet as this makes dissolving more difficult.
9. resuspend the pellet in 10 µl nuclease-free water by pipetting carefully.
- 10 µl purified ligation was mixed with 40 µl competent cells and electroporated into DH5alpha.
- 1 ml SOC medium (SOB medium + 20 mM glucose) was used and incubation was carried out for 1h at 37°C, shaking at 500 rpm in 2 ml eppis.
- Culture was then centrifuged at 4000 rpm for 5 minutes, and resolved in 100 µl medium.
- Whole sample was plated onto LB/agar plates supplemented with appropriate antibiotic and incubated o/n @ 37°C
Synthetic construct
Digest of synthetic construct and PCR-product (in pSB1C3)
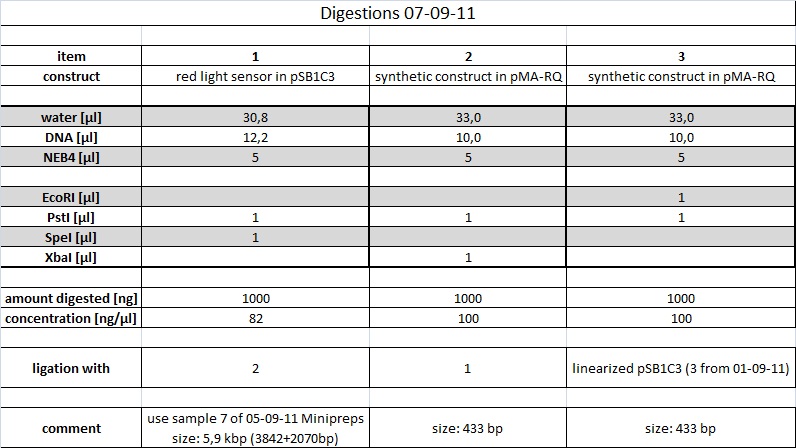
Samples were loaded on preparative gel (1 %, TAE 1x, 100 V, 2 h 30 min) and subsequently cut with a new scalpel for each sample using 365 nm illumination as short as possible. Gel slices were purified using Promega's Wizard Kit according to manufacturer's instructions. Picture was taken after cutting of gel slices in order to reduce illumination.
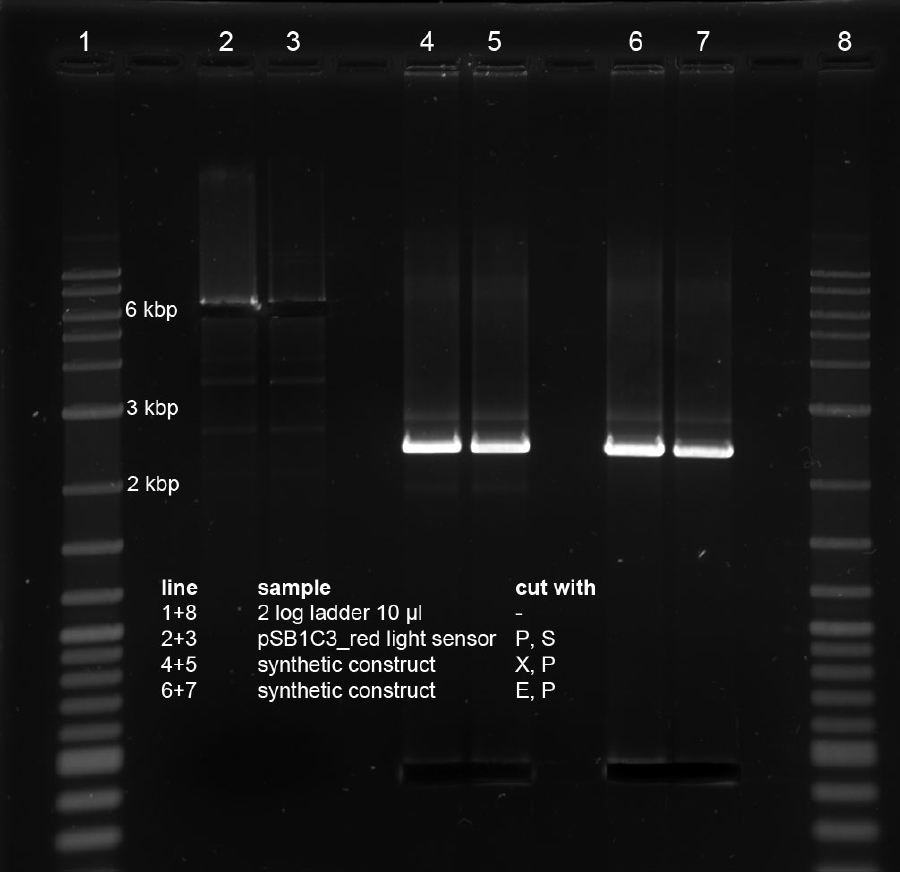
ligation of synthetic construct and PCR-product (in pSB1C3)
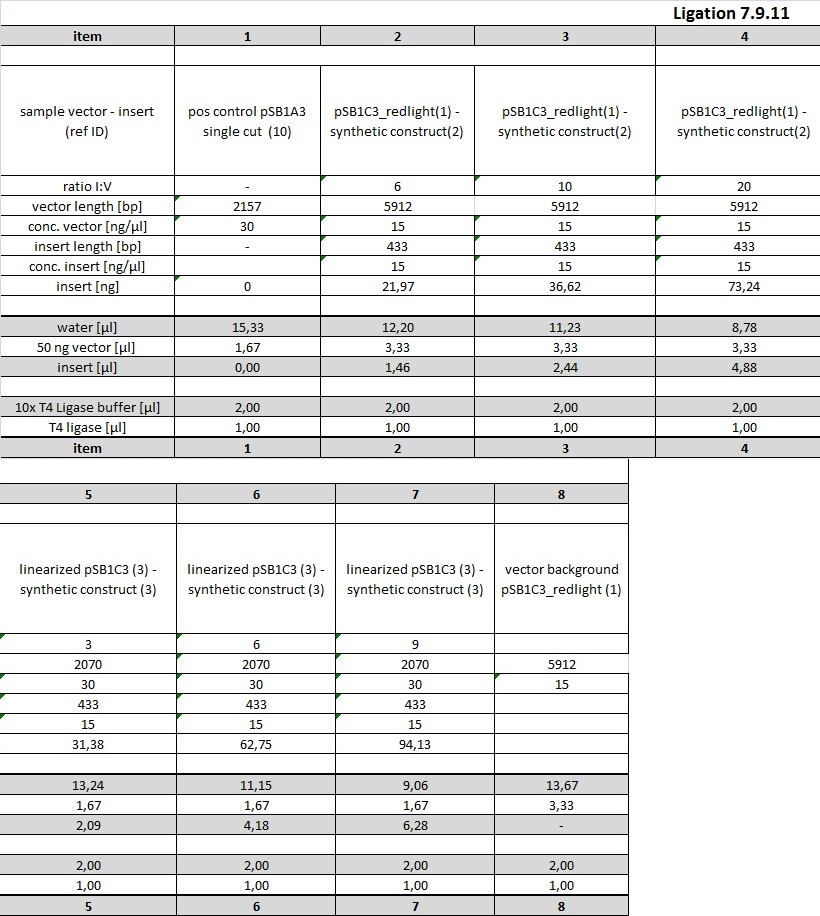
Ligation was performed at 16°C o/n
08-09-2011
People: Anna, Katharina
Cloning
AND-Gate plasmid
- DNA precipitation with glycogen/ethanol of the ligation samples from 7-9-11 accroding to protocol 7-9-11
- transformation via electroporation of 40 µl electro competent DH5alpha cells with the full volume of purified ligation samples from 7-9-11
Reporter plasmid and T7ptag
-overnight cultures for mini prep of transformations from 7-9-2011:
four clones of plates:
2 (T7prom(2)_RBS_lacZ(4))
3 (T7prom(2)_RBS_lacZ(4))
5 (RBS_lacZ(3)_T7prom(1))
6 (pSB1C3(3)_T7pol(5))
7 (pSB1C3(3)_T7pol(5))
one clone of plate 13 (synthetic construct in delivery vector pMA-RQ)
Other work
- production of LB-Agar plates with Kanamycin (500 ml), Chloramphenicol (300 ml) and Ampicillin (500 ml)
- overnight cultures of K322127 in DH5alpha (Cam, in the dark) and cpH8 inDH5alpha (Kan) for Miller Assay the next day
09-09-2011
People: Anna, Katharina, Thorsten
Results
Std Primer VR seems to misprime to B0015 double terminator: http://partsregistry.org/Help:Primers/Problems_using_VF2_and_VR_to_amplify_BioBrick_parts
Could be a problem when sequencing into our construct from downstream primer binding site as we use B0015 in synthetic construct. Because sequencing was done succesfully using this primer by others, we'll try it keeping the mispriming in mind. --> B0014 would be a better terminator for next time.
pSB1C3 is using B0056 as terminator downstream of suffix.
Cloning
Reporter plasmid and T7 polymerase
- Plasmid isolation -> Miniprep of the over night cultures from 8-9-11 according to manufacturers instructions from Metabion
- measurement of the DNA concentration of the following samples:
2a: 92 ng/µl
2b: 122 ng/µl
2c: 131 ng/µl
3a: 98 ng/µl
3b: 152 ng/µl
3c: 124 ng/µl
3d: 112 ng/µl
5a: 52.5 ng/µl
5b: 55 ng/µl
5c: 74.5 ng/µl
5d: 87 ng/µl
6a: 134 ng/µl
6b: 118 ng/µl
6c: 101 ng/µl
6d: 140 ng/µl
7a: 76.5 ng/µl
7b: 113 ng/µl
7c: 83 ng/µl
7d: 71.5 ng/µl
13: 79 ng/µl
- control restriction digest of the 21 DNA samples (2a-d, 3a-d, 5a-d, 6a-d, 7a-d,13) with the restricition enzymes E,P
total volume 50 µl (39 µl nf H20, 5 µl DNA, 5 µl NEB4 Buffer, 0,5 µl of each enzyme)
incubation for 1 hour at 37°C, inactivation for 20 min at 80°C
- analytical gelelectrophoresis ( 1%, 1x TAE, 12 wells, 120 V, 1 hour 30 minutes)
50 µl DNA + 10 µl loading buffer -> 40 µl sample in each well
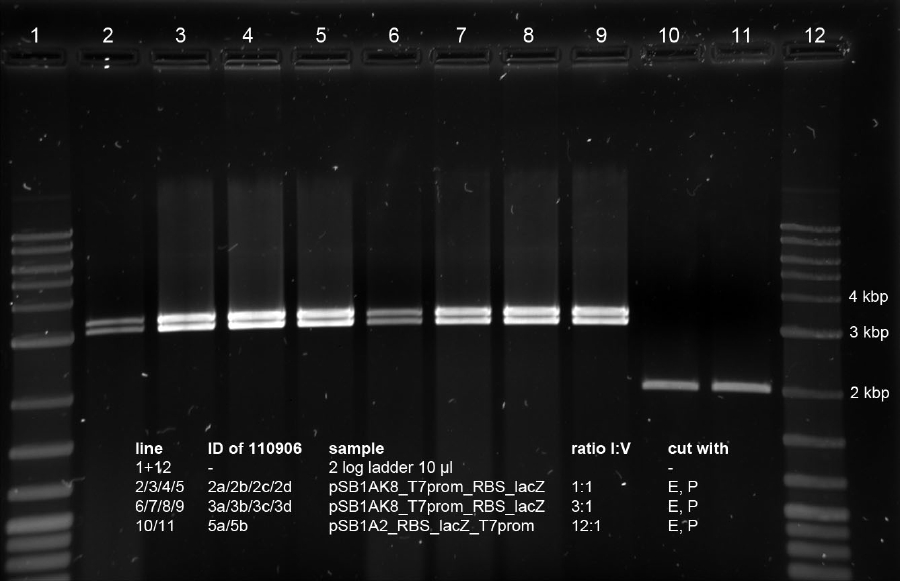
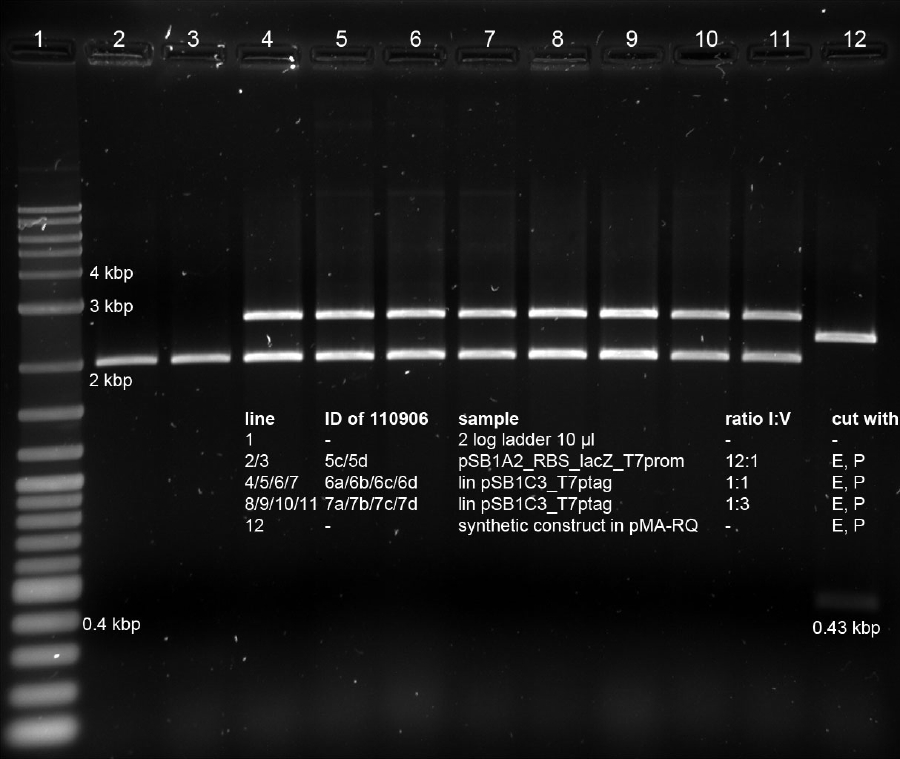
Size of T7promotor with RBS_lacZ looks correct (RBS_lacZ approx 3kbp pSB1AK8 approx 3.4 kbp). Ligation of 46 bp T7promotor into pSB1A2_RBS_lacZ (ID 5) didnt yield any correct clones, only vector background could be picked. Samples were discarded. Transformation of sample 4 resulted in short-circuit and was lost. Ligation of T7ptag into pSB1C3 looks fine. (T7ptag approx 2.6 kbp)
AND-Gate plasmid
transformation output: vector background looks fine
following clones have been picked:
Other Work
Next steps
Besides ligation of T7ptag insert, pSB1C3_T7ptag vector will be opened to ligate with red light_synthetic construct insert--> see 10-09-11.
Send clones for sequencing on Monday.
Testing
Miller Assay
two cultures in LB medium:
K322127 in DH5alpha with three incubation conditions: lighted, dark and under off-light
cpH8 in DH5 alpha as a control
conditions of bacterial growth: 37 °C, shaken, culture K322127 in DH5alpha grown in the dark prior to the experiment
volume of bacterial suspension used: 500 µl
incubation time for Miller Assay: 10 min (37 °C)
improvement to prior experiments: start OD between 0,15 and 0,17, correct antibiotics applied, incubation time of Miller Assay only 10 min.
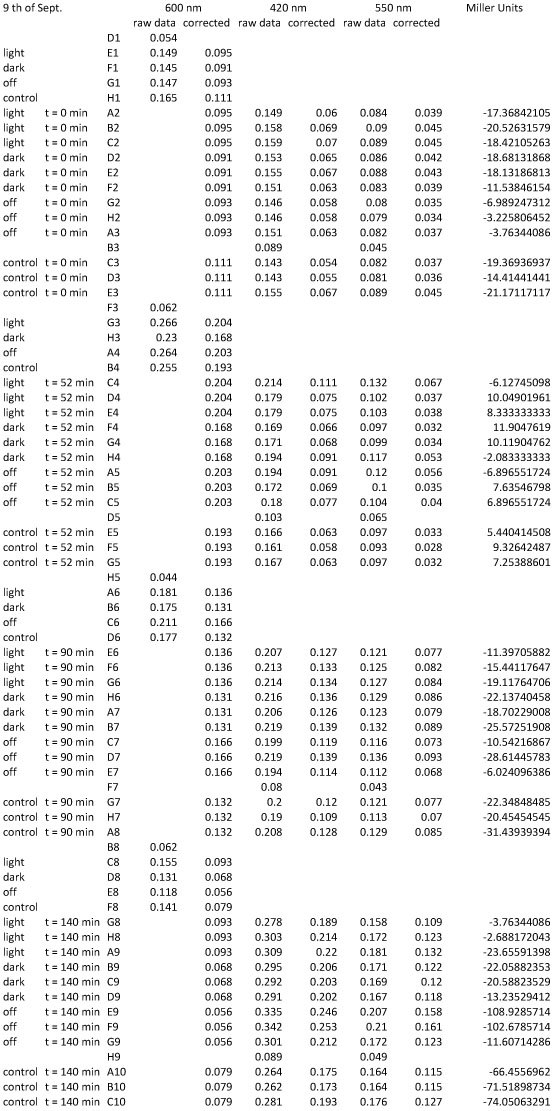
Results
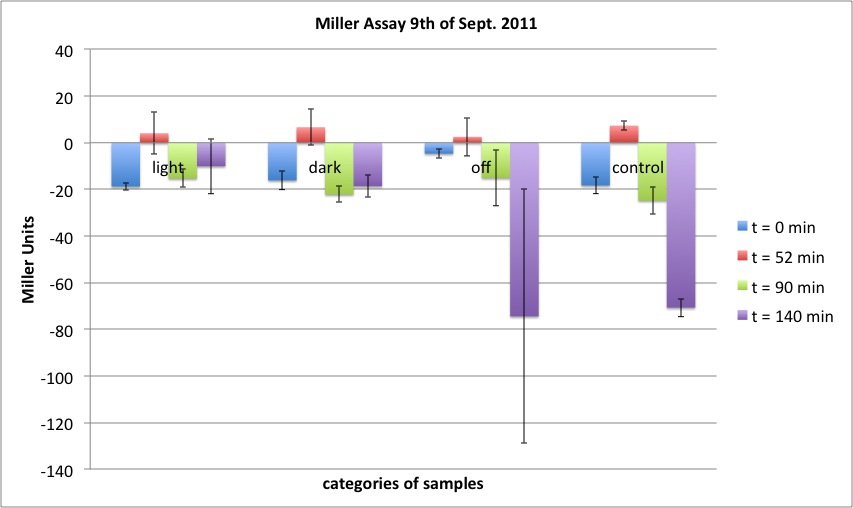
10-09-2011
People: Susan, Wolfgang, Thorsten
Cloning
AND-Gate plasmid
red_light sensor - synthetic construct ligation check
- Plasmid isolation -> Miniprep of the over night cultures from 09-09-11 according to manufacturers instructions from Metabion. 4 ml culture was used and 1 ml culture was saved for potential glycerol stocks
- control restriction digest of the 21 DNA samples (2a-d, 3a-d, 5a-d, 6a-d, 7a-d,13) with the restricition enzymes E,S
total volume 10 µl (7 µl nf H20, 1 µl DNA, 1 µl NEB4 Buffer, 1 µl of each enzyme)
nuke for 1 minute at 800 W (microwave)
- analytical gelelectrophoresis ( 0.7%, 1 x TAE, 12 wells, 160 V, 20 minutes)
10 µl DNA + 2 µl loading buffer -> 12 µl sample in each well
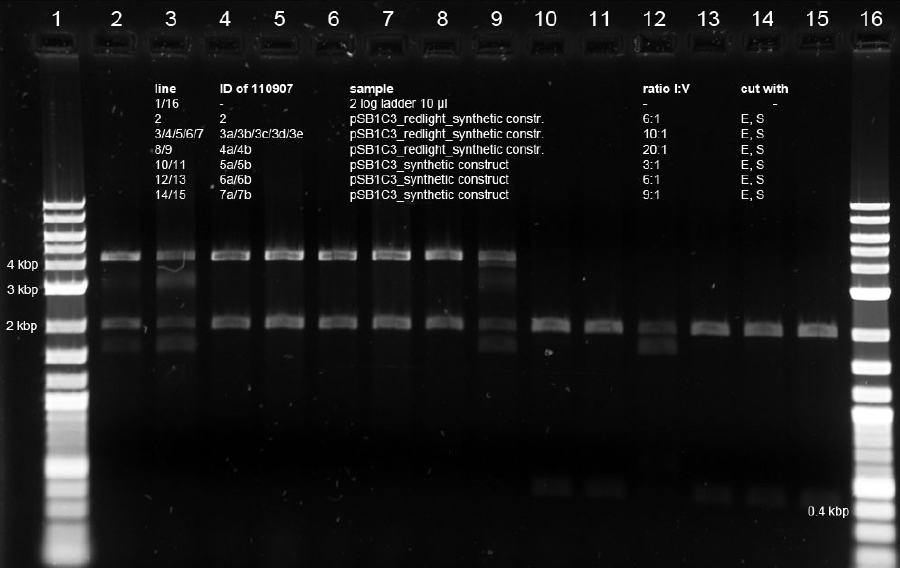
Sample ID 3c was choosen to be used for further cloning. pSB1C3 with red light sensor and synthetic construct
Nanodrop derived DNA conc. of 3c: 114 ng/µl
samples 2, 3a, 4b and 6a were discarded
sample 7b was choosen to be synthetic construct in pSB1C3 --> sequencing on monday
last step: assembly of redlight sensor_synthetic construct in pSB1C3 with T7ptag
we took 2 approaches: ligation of T7ptag as insert and vector, respectively
Digestions
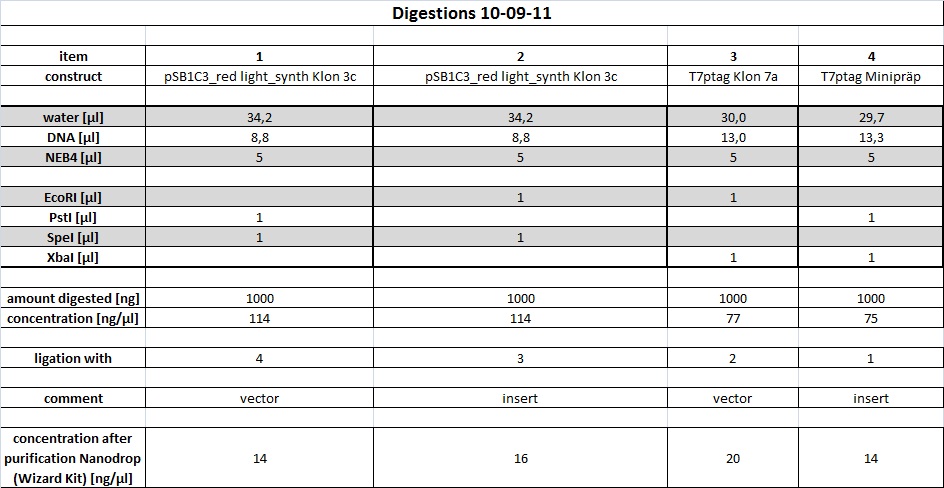
samples were loaded on 1 % preparative agarose gel and run for 1.5 h at 120V
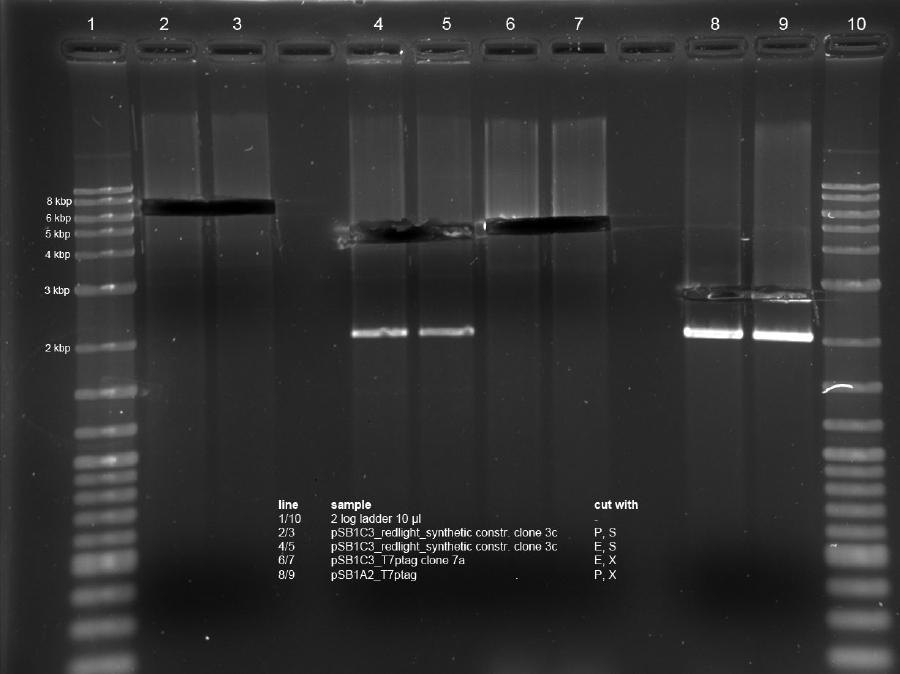
Samples were cut (365 nm illumination as short as possible, picture taken after cutting of gel slices) and purified using Promegas Wizard purification kit according to manufacturers instructions.
Ligations
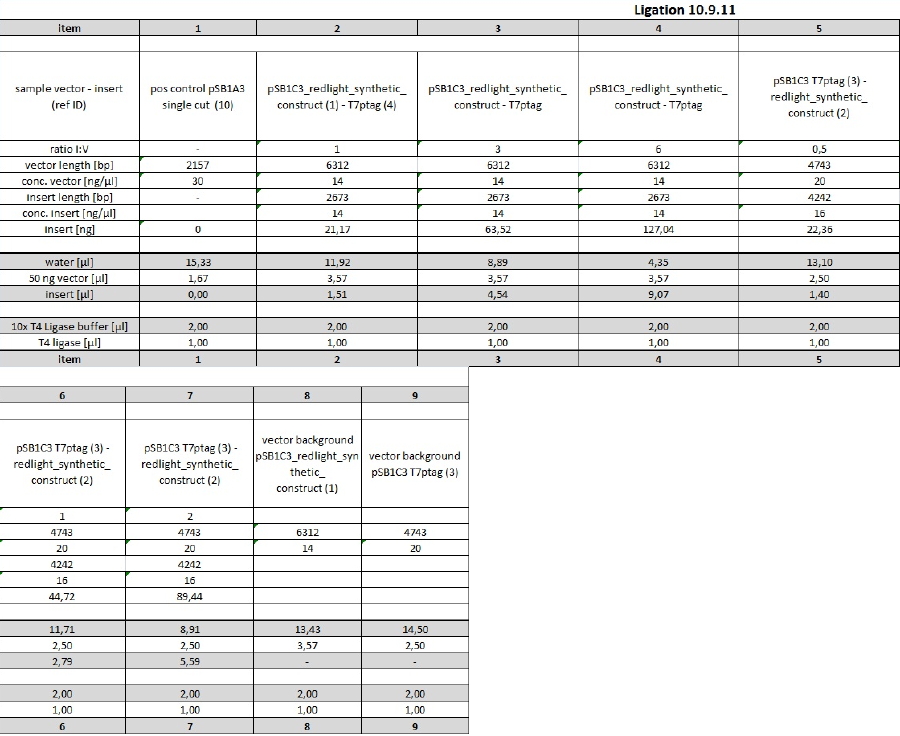
Samples were incubated o/n at 16°C.
11-09-2011
People: Katharina, Flo, Anna
Cloning
Glycogen/EtOH-precipitation of ligation samples from 10-09-11 and transformation
Put everything on ice before starting to work! Pre-cool centrifuge!
See protocol for precipitation/transformation from 07-09-11[See Methods]
Changes made:
- all solutions were icecold
- pellet was air-dryed for about 10 min
- autoclaved water was used instead of nuclease-free, due to quick subsequent use in the transformation
- resuspending of the bacterial pellet after transformation was difficult (tried pipetting and vortexing), because the wrong centrifuge has been used
- 1,5 ml-Eppis were used instead of 2 ml-Eppis for the shaking step at 37°C in SOC
Samples 1, 8 and 9: comp. cells used were DH5a from 01-07-11
Samples 2 - 7: comp. cells used were DH5a from 30-08-11
Antibiotic resistance:
- 1: Amp
- 2 - 9: Cam
Testing
Inoculation of o/n-cultures for tomorrow´s Miller Assay
10 ml LB+antibiotic in 50 ml-falcon tube
add 10 ul (= 1:1000 dilution) of yesterday´s o/n-cultures:
- bluelight + lacZ in DH5a (Amp)
- redlight + lacZ (trunc.) in DH5a (Cam)
- neg. contr. Cph8 in DH5a (Kan)
Wrap falcons with aluminium foil, so growth can occur in the dark!
Shake o/n @ 37°C, 180 rpm
Miller Assay with blue light construct
two cultures in M9 medium:
blue light - lacZ on pSB1A2 in DH5alpha with two incubation conditions: lighted in blue light and dark
cpH8 in DH5 alpha as a control under room light
conditions of bacterial growth: 23,5 °C, shaken, both cultures grown under room light prior to the experiment
volume of bacterial suspension used: 0,1 ml
incubation time for Miller Assay: 5 min (37 °C)
improvement to prior experiments: start OD between 0,15 and 0,17, correct antibiotics applied
Start of Assay:
- measurement of Abs(600nm)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood)
- 0,1 ml of the samples are taken and transferred to the Zbuffer
- mix the solution by vortexing for 10 s ( all samples equal vortexing time)
- transferring of 100 μl supernatant to 96 well plate (for photometer)
- initiation of assay with 20 μl of ONPG (4mg/ml) NOTE START TIME
- incubation at 37 °C
- stop reaction with 50 μl of 1 M Na2CO3 NOTE STOP TIME
- measurement of Abs (420nm) and Abs (550nm)
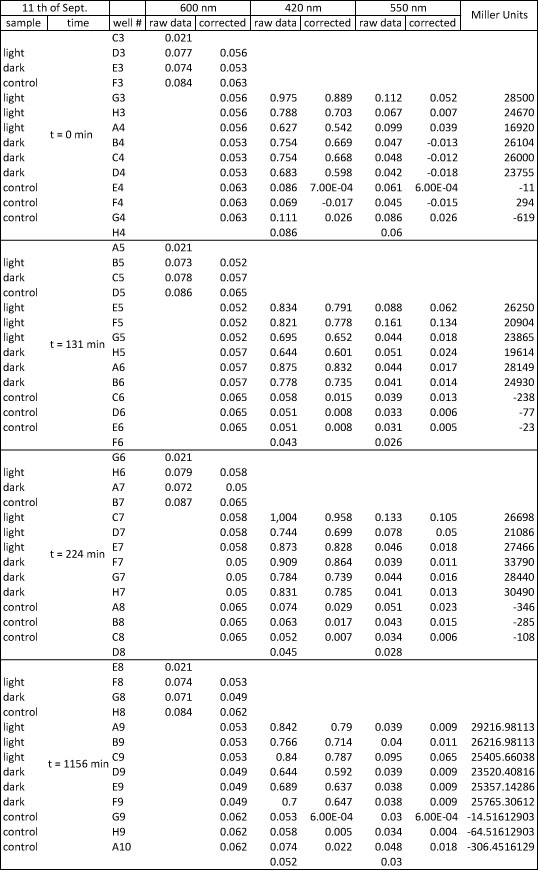
Results
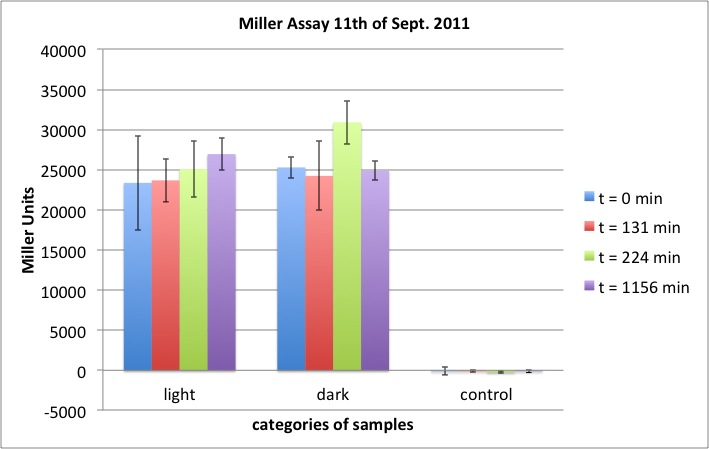
Problems: 1. cells had a party over night: three LED (blue on, red on, red off) were on alternatively 2. did cells really grow?
12-09-2011
People: Thorsten, Anna
Cloning
Part I746907 (T7 promoter, GFP) in pSB1A2 was electroporated into DH5alpha, and plated onto a LB-Agar Amp plate. This part can be used as a reporter plasmid for our AND-Gate in e. coli CP919.
AND-Gate: Picking of colonies
Transformation and ligation efficiency of control was several thousand clones. Vector controls were lawn due to usage of CAM-plasmid contaminated electorcompetent DH5 alpha. All contaminated DH5alpha from 01-07-11 have been discarded.
3 clones of each plate have been picked and inoculated in 5 ml LB/cam o/n.
Other Work
sequencing of clones
Sequence of Blue light promotor with downstream RBS+lacZ is correct.
Sequence of first 1000 bp of PCR-Product shows one possible silent mutation in PcyA (Phycocyanobilin:ferredoxin oxidoreductase) gen on Position 45 T->C. Resulting triplet is CCC instead of CCT, which both code for proline.
Rest of PCR product should be sequenced aswell? Two more primers would be necessary to cover whole sequence.
Or: functionality check using CP919 strain should result in comparable Miller Unit output between PCR product only and red light original part K322127 as CP919 is Φ(OmpC-lacZ)10-25
Testing
Miller Assay with blue light construct
two cultures in LB medium:
blue light - lacZ on pSB1A2 in DH5alpha with three incubation conditions: lighted in room light, dark and blue light
cpH8 in DH5 alpha as a control under room light
conditions of bacterial growth: 23,5 °C, shaken, both cultures grown in the dark prior to the experiment, two hours before the experiments: cultures were subdivided and lit as described
volume of bacterial suspension used: 50 µl (or dilutions with LB medium)
incubation time for Miller Assay: 5 min (37 °C)
Start of Assay:
- measurement of Abs(600nm)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood) in 2 ml tube
- 50 μl (as a dilution 25 µl + 25 µl LB medium or 10 μl + 40 μl LB medium) of the samples are taken and transferred to the Zbuffer
- mix the solution by vortexing for 10 s (all samples with equal vortexing time)
- transferring of 100 μl supernatant to 96 well plate (for photometer)
- initiation of assay with 20 μl of ONPG (4mg/ml) = START
- incubation at 37 °C
- stop reaction with 50 μl of 1 M Na2CO3 = STOP after exactely 5 min
- measurement of Abs (420nm) and Abs (550nm)
- when solution was too yellow (OD > 2.0), stopped mixtures were diluted 1 : 7 in dd H2O
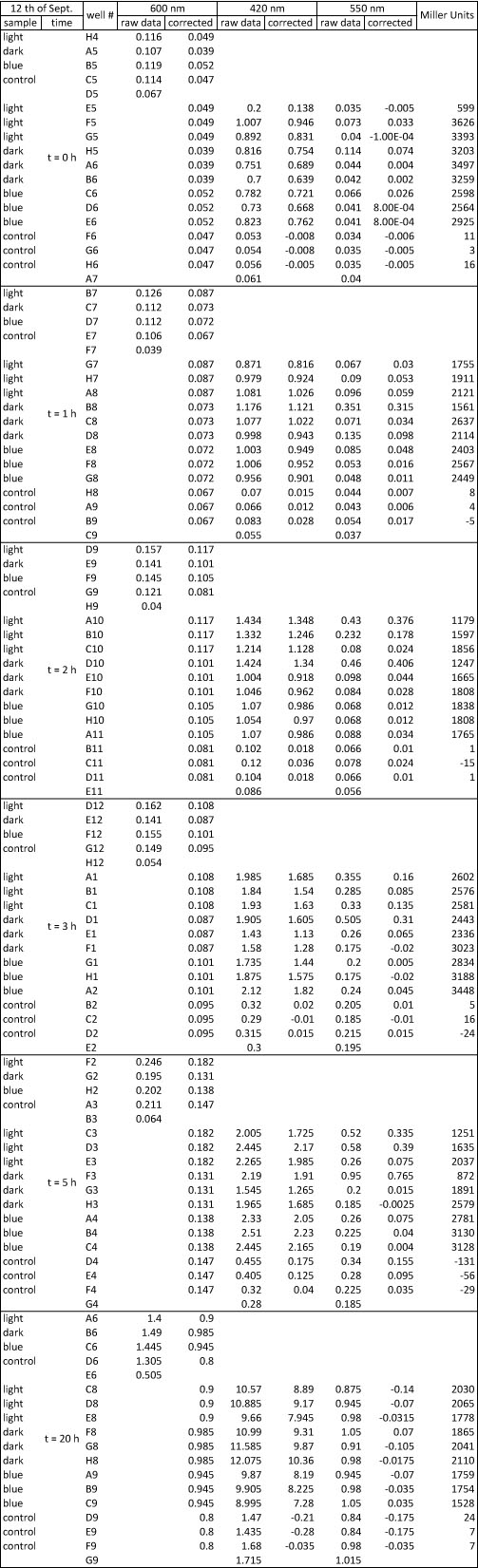
Results
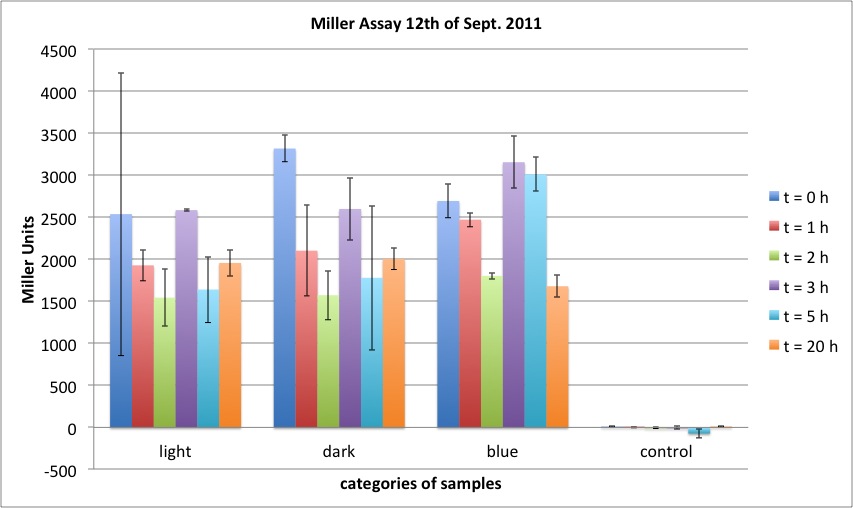
Problems: 1. did cells grow up to 20 h and act normally?
13-09-2011
People: Simon, Anna, Alex, Thorsten
Cloning
One colony from yesterday's transformation of Part I746907 in pSB1A2 was picked and inoculated into 5 ml LB Amp. Incubation o/n at 37 °C
AND-Gate
Minipreps of picked cones on 12-09-11 were done according to Metabion's instructions. Concentrations:
2a: c = 68.5 ng/µl
2b: c = 68.0 ng/µl
3a: c = 118 ng/µl
3b: c = 65.0 ng/µl
5a: c = 122 ng/µl
5b: c = 71.5 ng/µl
6a: c = 122 ng/µl
6b: c = 119 ng/µl
6c: c = 51.0 ng/µl
After restriction digest using E and P, samples were separated on a 0.8 % gel:
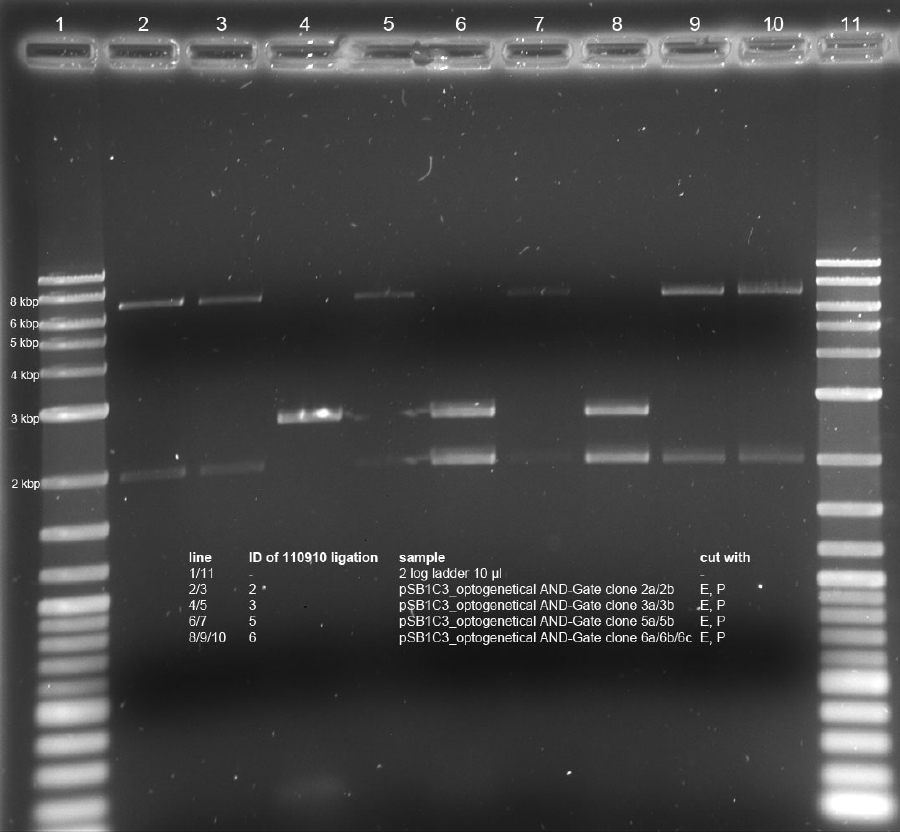
Vector prep pSB1A3 and pSB1C3
- The restricted vectors ID10 pSB1A3 and ID12 pSB1C3 from 30-08-2011 were applied onto a 1 % agarose gel and prepped using the Wizard SV gel purification kit.
- The vector pSB1A3 was single cut, while pSB1C3 was cut with E and P (from K322127). The expected lengths are about 2 kbp for pSB1C3 and about 3 kbp for pSB1A3, as it also contains a RFP cassette.
- The measured concentrations were 12.5 ng/µl for pSB1A3 and 11.5 ng/µl for pSB1C3, in a resuspended volume of 50 µl.
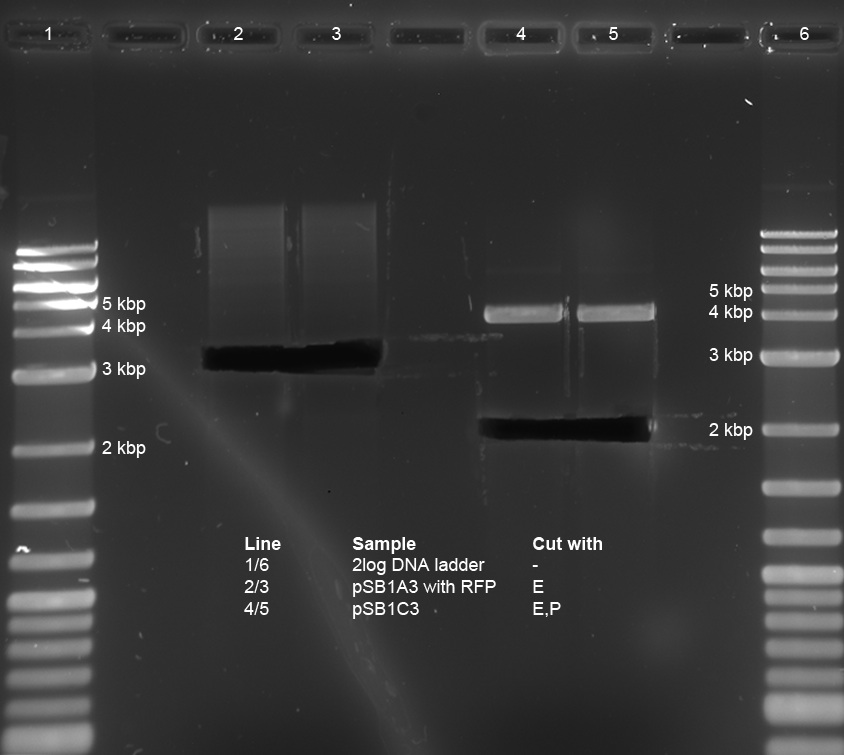
Testing
no Miller Assay today
Inoculation of o/n-cultures for tomorrow´s Miller Assay
10 ml LB+antibiotic in 50 ml-falcon tube
add 20 µl (= 1:1000 dilution) of yesterday´s o/n-cultures:
in LB medium:
- bluelight + lacZ in DH5a (Amp)
- neg. contr. Cph8 in DH5a (Kan)
in M9 medium:
- redlight + lacZ (trunc.) in DH5a (Cam)
- neg. contr. Cph8 in DH5a (Kan)
Wrap falcons with aluminium foil, so growth can occur in the dark!
Shake o/n @ 37°C, 180 rpm
Other Work
1. preparation of Zbuffer and Na2CO3 solution for next day
2. Transformations:
- chemical transformation of pSB1AK8 T7prom RBS lacZ (plasmid from clone 2c, 09.09.2011) in BL21 DE3. The BL21 cells we received from Lars Mitschke turned out to be chemically competent cells. Transformation was performed based on a protocol from openwetware.org. For detailed description of the protocoll see Methods.
- electroporation of Part R2 (= RBS + lacZ) in pSB1A2 using today's and the old electrocompetent DH5alpha cells.
Sequencing results
pSB1AK8_T7prom_RBS_lacZ_VF2_110909_clone_2c-GATC-VF2-545457: Sequence shows one missing T in scar between T7promoter and RBS of lacZ. Rest of sequence is fine.
Electrocompetent cells
- new electrocompetent cells (DH5alpha) were made by the following protocol:
- 20 ml LB medium was inoculated with 1 µl electrocompetent DH5alpha (kindly provided by Andrea Mückl)
- 350 ml LB medium were inoculated with 15 ml of the overnight culture and incubated at 37°C until a OD(600) of 0,7 was reached
- the medium was distributed on falcons and the cells were centrifuged at 4500 rpm for 20 min at 4°C
- after discarding the supernatant 350 ml of sterile ice-cold 10% glycerol was added and the pellets were resuspended using a pipet
- the resuspension was centrifuged at 4500 rpm at 4°C for 11 minutes
- after discarding the supernatant the cells were resuspended again in 350 ml of ice-cold 10% glycerol (cells kept chilled all the time)
- and again centrifuged at 4500 rpm at 4°C for 11 minutes
- the supernatant was discarded and the cells were resuspended in the remaining supernatant
- the cells were dispensed in 40 µl aliquots and stored at -80°C for further use
14-09-2011
People: Simon, Anna, Alex, Thorsten
Cloning
The over night culture of part I746907 (pSB1A2) in DH5alpha was centrifuged and the supernatant was removed. Because of time reasons, the miniprep will be done tomorrow, the pellet was stored at -20°C.
Furthermore, parts BBa_K568002 (blue light sensor, RBS, LacZ) and BBa_K568003 (T7 promoter, RBS, LacZ) need to be transfered into shipping vector pSB1C3. Therefore, the parts are digested, the vector has already been cut with E and P.
Digests
Blue light sensor, RBS, LacZ in pSB1A2 (tube labelled: Miniprep 04-09-2011, clone 2, c=50 ng/µl) and T7 promoter, RBS, lacZ in pSB1AK8 (miniprep from 09-09-2011, clone 2c, c=131 ng/µl) were cut with E and P.
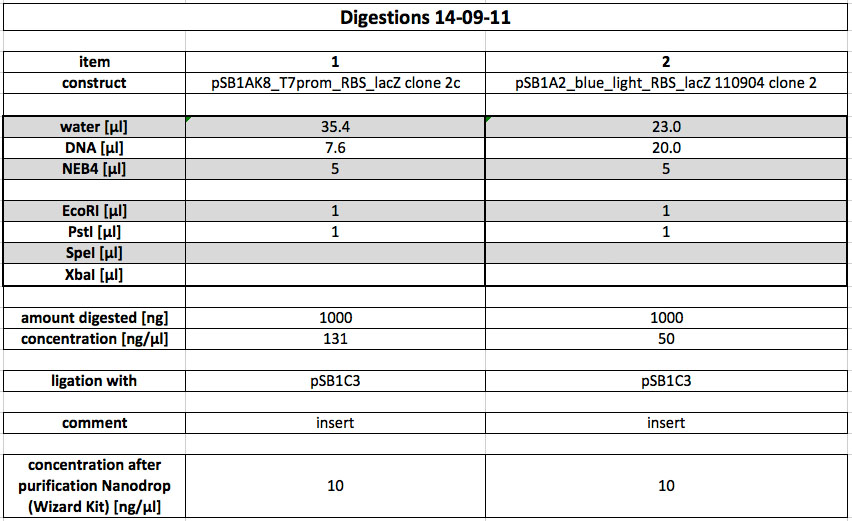
Next, the digested DNA was loaded onto a preparative 1% gel. After the electrophoresis was complete, the correct bands were cut out on the UV transilluminator (365 nm) before the following gel photo was recorded:
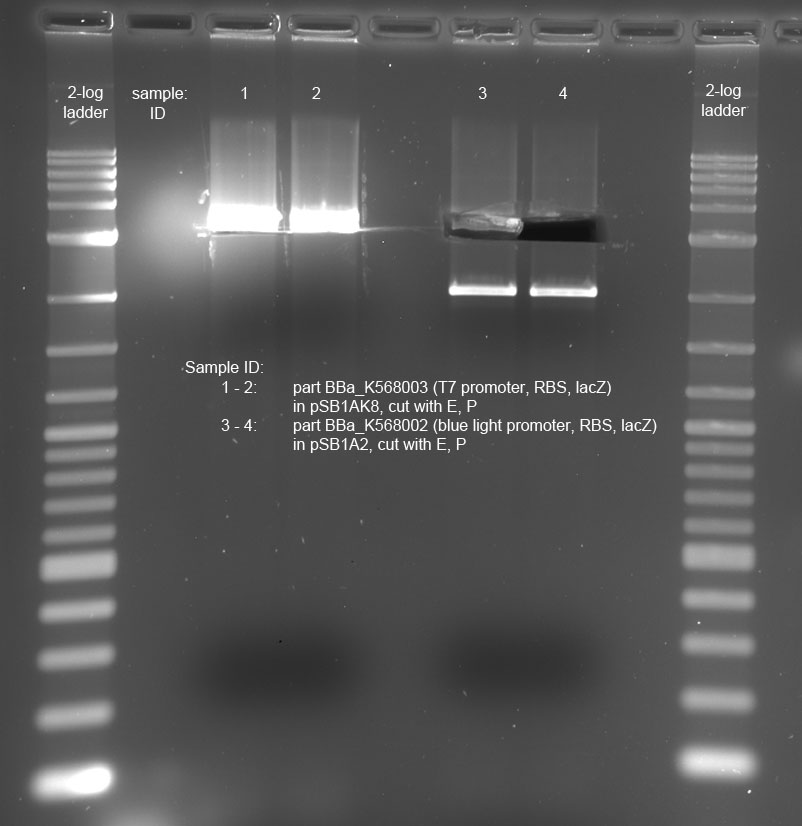
The gel looks fine. Cutting out the correct band had to be done very carefully in lanes 1 and 2, because the bands for the vector and the insert were very close together (vector: 3.4 kb, insert: 3.1 kb). Lanes 3 and 4 also show the expected bands, at about 3.2 kb for the insert and about 2 kb for the vector.
Finally, the DNA was extracted and purified from the cut-out gel slices using the Promega Wizard Kit according to the kit protocol.
The following concentrations were measured (Nanodrop):
blue light sensor, RBS, lacZ: 10 ng/µl
T7 promoter, RBS, lacZ: 10 ng/µl
Ligations
As a next step, the following ligation was performed (see table below). We used the opportunity to compare the old (NEB) and the new (Metabion) ligase. The ligation was incubated at 16 °C over night.
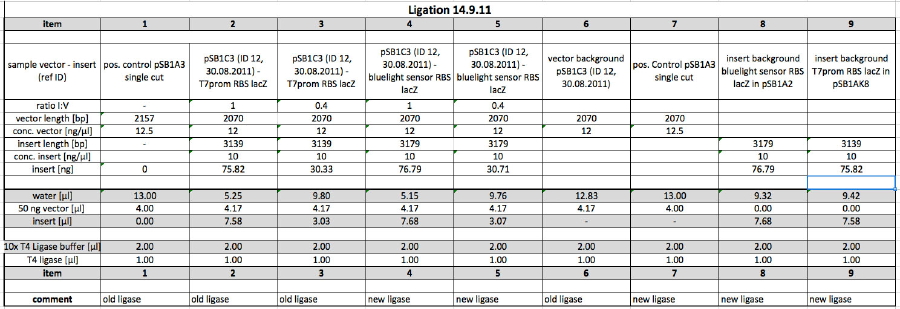
Testing
Miller Assay with blue light construct
two cultures in LB medium:
- bluelight + lacZ in DH5a (Amp), two temperatures (30 °C and RT = 22,8 °C), three lightning conditions (room light, dark and blue light)
- neg. contr. Cph8 in DH5a (Kan) at 30 °C
volume of bacterial suspension used: 0,1 ml
Start of Assay:
- measurement of Abs(600nm)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood) + sample
- 0,1 ml of the samples are taken and transferred to the Zbuffer
- mix the solution by vortexing for 10 s (all samples equal vortexing time)
- transferring of 100 μl supernatant to 96 well plate (for photometer)
- initiation of assay with 20 μl of ONPG (4mg/ml) NOTE START TIME
- incubation at 37 °C, 4 or 5 minutes
- stop reaction with 50 μl of 1 M Na2CO3 NOTE STOP TIME
- measurement of Abs (420nm) and Abs (550nm)
Results
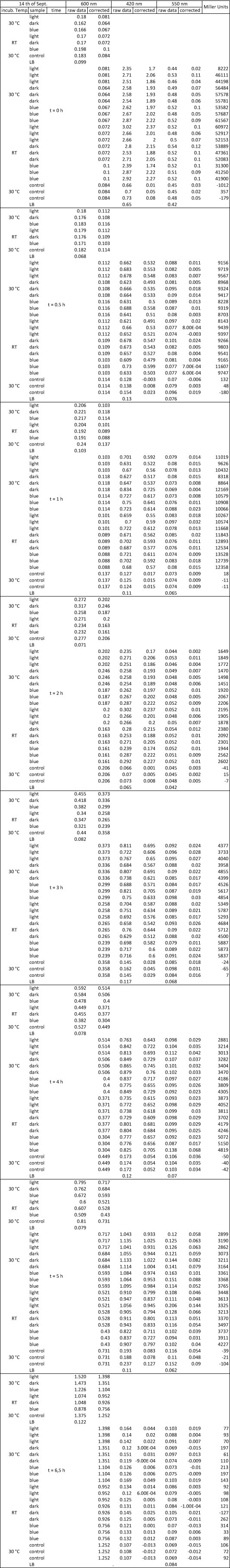
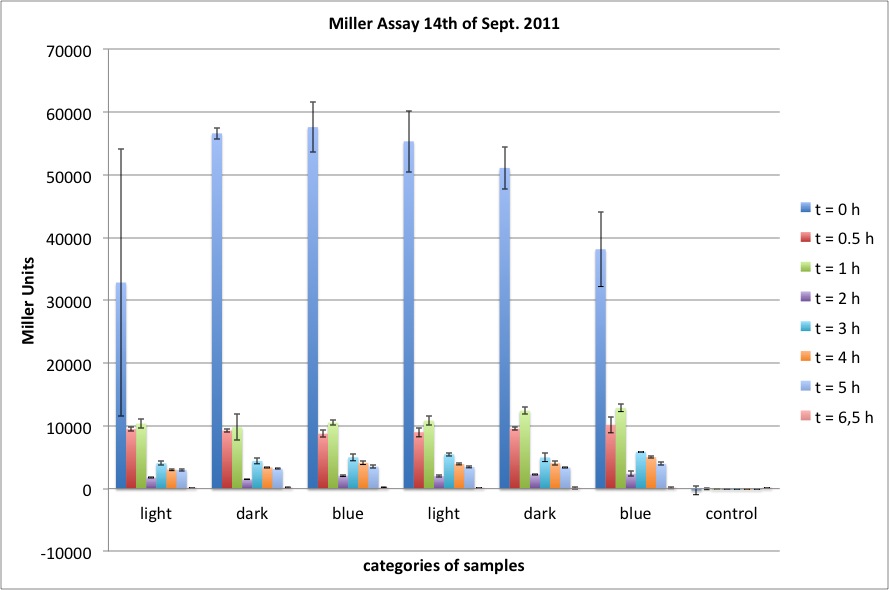
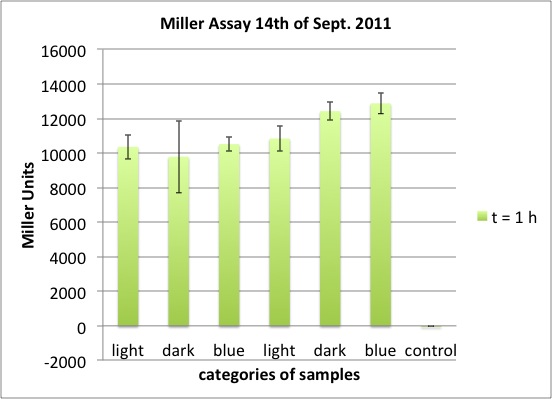
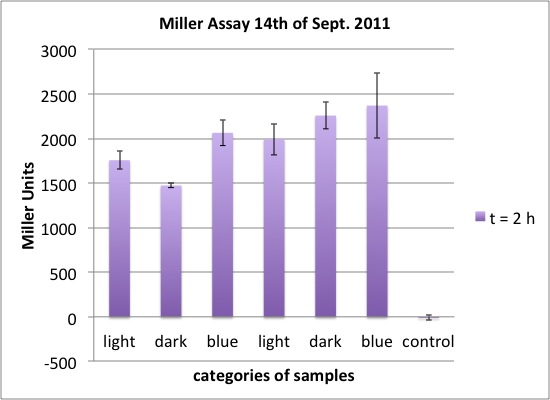
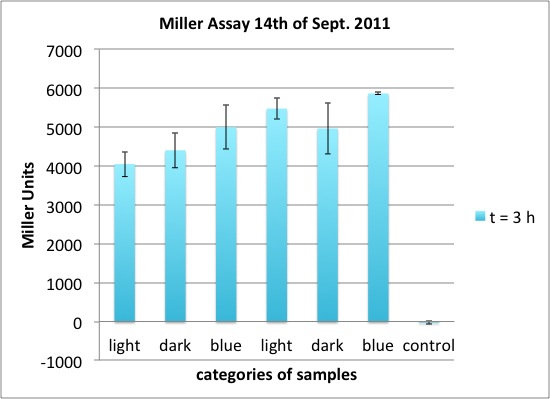
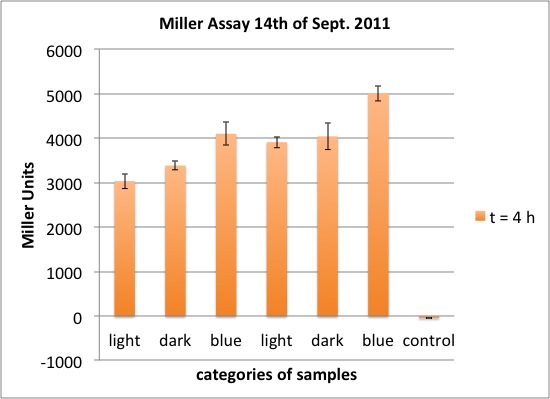
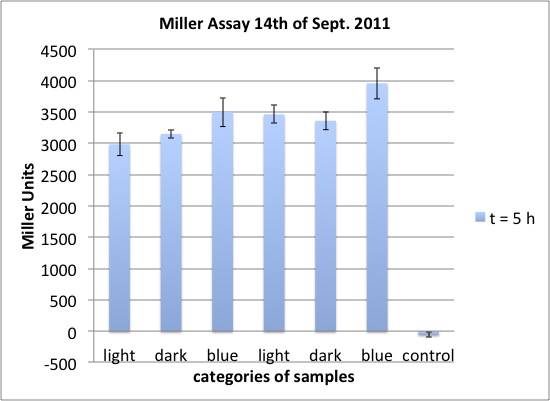
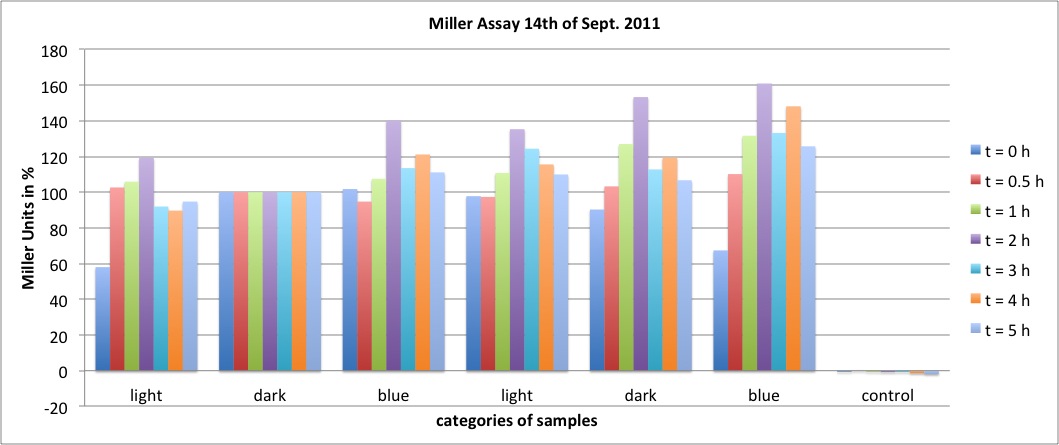
Is the spectrometer linear over such a long range of OD's?
no Miller Assay with red light sensor as cells didn't grow:
in M9 medium:
- redlight + lacZ (trunc.) in DH5a (Cam)
- neg. contr. Cph8 in DH5a (Kan)
15-09-2011
People: Simon, Anna, Alex
Cloning
Miniprep
Part I746907 (pSB1A2) was purified from yesterday's DH5alpha-pellet using the miniprep-kit according to protocol. The concentration was determined to be 63 ng/µl using Nanodrop.
Glycogen/Ethanol precipitation and Transformation
Yesterday's ligations were purified using glycogen/ethanol precipitation. The yielded 10 µl DNA-Solution were used completely for Transformation in DH5alpha. After electroporation, the cells were incubated at 37 °C for 1h in SOC-media. After this, they were plated onto LB-Agar plates with the appropriate antibiotics.
Digestions
The cloning of our last unfinished part (BBa_K568004), "optogenetical AND-Gate without first promoter - choose your first input" was started today. The part consists of our synthetic construct plus t7ptag. Our synthetic construct (in pMA-RQ) was digested for preparation using E and S, while the miniprepped part I746907 was digested using E and S to check if the prepped plasmids are correct (a mistake was made during preparation of the digest, so a second digest was made at the same time. Both digests were loaded onto the analytical gel):
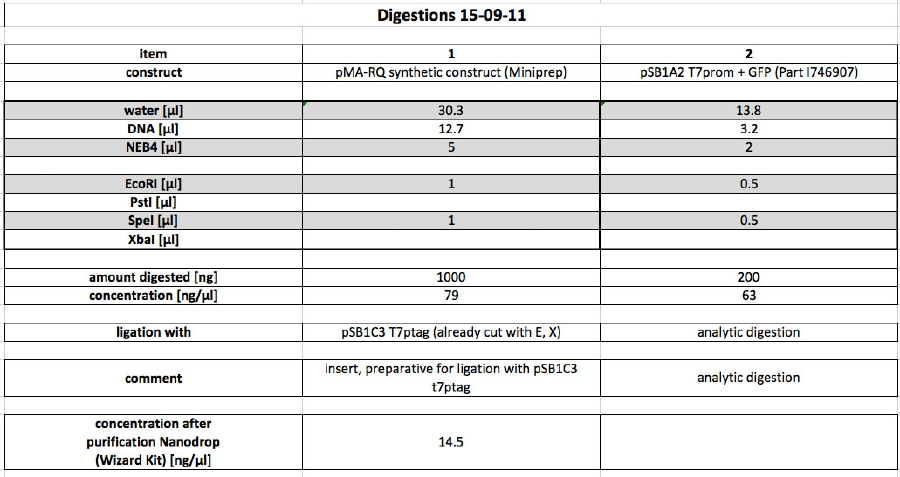
The product of the preparative digest was loaded onto a preparative 1% agarose gel. After electrophoresis, the desired band was cut out on the UV-transilluminator (365 nm) before the gel photo was recorded:
The product of the preparative digest was loaded onto a preparative 1 % agarose gel. After electrophoresis, the desired band was cut out on the UV-transilluminator (365 nm) before the gel photo was recorded: :

The gel looks as expected. The cut out bands around 400 bp are the synthetic construct while the vector can be seen at 2.4 kb.
The product of the control digest was loaded onto an analytic 0.8 % agarose gel. The following gel photo was recorded:

Lane 1 (correct digest): Part BBa_I746907 is 924 bp long, the gel shows the corresponding bands. The vector pSB1A2 is 2079 bp long and can also be seen at the expected spot.
Lane 2 (wrong digest): The bands show that the digest doesn't work properly in 10x NEB4 buffer. The band around 3 kb is uncut or singly cut vector with insert.
Testing
Miller Assay with reporter construct
We incubated two cultures over night in LB medium with the appropriate antibiotics:
- BBa_K568003 (T7 promotor and β-galactosidase) in BL21(DE3);
- Negative control BBa_I732017 (rbs and β-galactosidase) in DH5 alpha.
The cultures were diluted to a OD_600 of about 0.7 and incubated for another 30 min. Then, the cultures were induced with IPTG at different concentrations:
- BBa_K568003: 0 mM, 0.01 mM, 0.1 mM, 1.0 mM and 1.5 mM;
- BBa_I732017: 0 mM and 1.0 mM.
Before induction and every 30 min, the Miller Units were measured as follows:
- measurement of Abs(600nm) in plate reader (volume of bacterial suspension should be equal in all wells, ideally 170 µl)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS + 40 μl of Chloroform (under fume hood) + 500 µl sample solution (e.g. 430 µl medium + 20 µl bacterial suspension)
- mix the solution by vortexing for 10 s (all samples with equal vortexing time)
- let chloroform settle down (this takes about 5 min, if tube still contains blurred solution, centrifuge for 1 min, RT, 4000 rcf)
- transfer 100 μl of supernatant to 96 well plate (for photometer)
- initiation of assay with 20 μl of ONPG (4mg/ml), mix well, NOTE START TIME
- incubation at 37 °C
- stop reaction with 50 μl of 1 M Na2CO3, mix well, NOTE STOP TIME
- measurement of Abs (420nm) and Abs (550nm)
The Miller Units were calculated according to the following equation:
Miller Units = 1000 * (ABS420 - (1.75 * ABS550)) / (time [min] * volume [ml] * ABS600)
The reaction time was 5 min and the culture volume used was 10 µl.
Other Work
Other Work
Testing the spectrometer (at the wavelengths 420 and 600 nm, because they are used in the Miller Assay: over which range are the OD values linear?)
samples:
- for 600 nm measurements: 170 µl of a bacterial culture in LB medium, blank with fresh LB medium, dilutions in LB medium (minor problem: cells could still grow during the experiment, especially as the plate reader heated the plate)
- for 420 nm measurements: 130 µl of one sample of Miller Assay from 13-09-2011 (had already been developed/stopped and stored o/n at RT), blank with ddH2O, dilutions in ddH2O
Results
Miller Assay Results
The Miller Assay yielded the following data set:
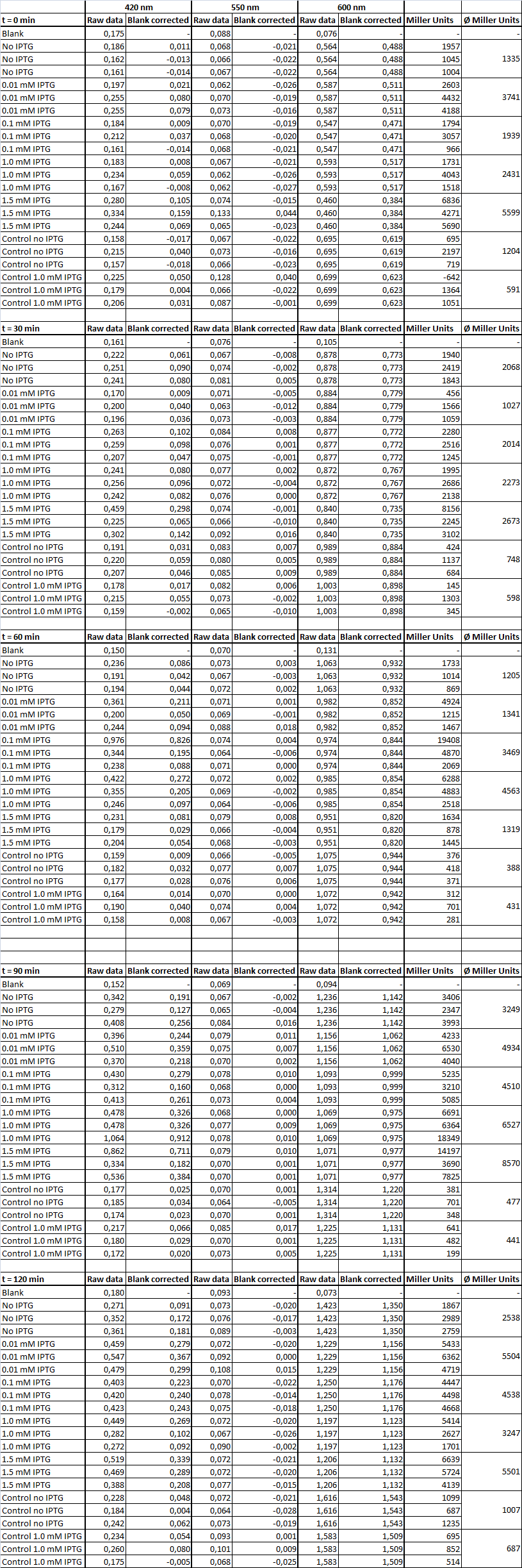
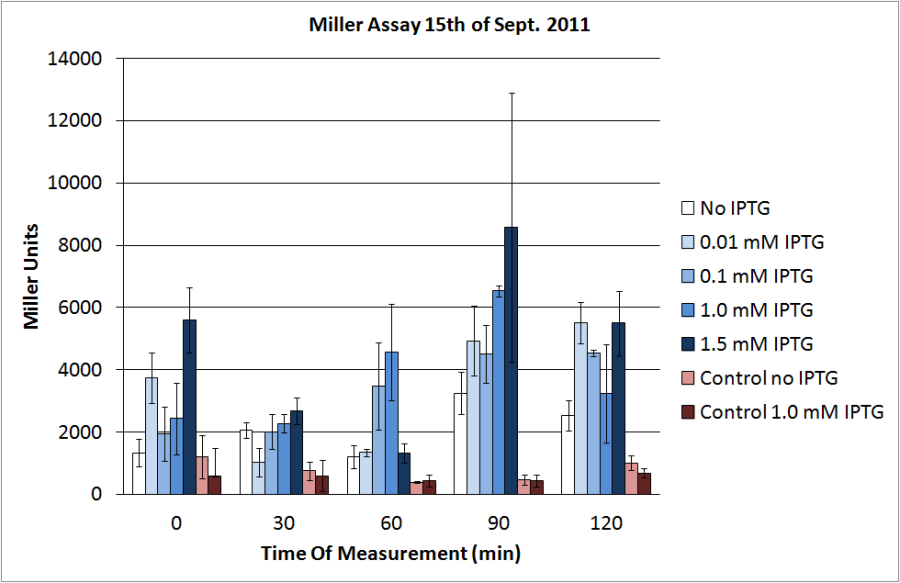
The data shows that the cultures induced by isopropyl-β-D-1-thiogalactopyranoside (IPTG) yield higher Miller Units than the uninduced control after 60 min. Therefore, a higher amount of β-galactosidase was produced. This shows that the part is working as expected. Furthermore a slight quantitative dependence of the Miller Units on the concentration of IPTG can be seen. After 120 min the Miller Units of the culture induced with 1.0 mM IPTG go down again, which might be evidence for increasing of proteolysis caused by death of bacteria due to a too high concentration of IPTG. It can also be observed that the uninduced control yields higher Miller Units than the β-galactosidase negative, induced control. This can be explained by leaky biosynthesis of the genome-coded T7 polymerase. If the production of T7 polymerase is more tightly controlled, the level of expressed β-galactosidase should go down to the minimum of the negative, induced control.
Testing of plate reader photometer
linear over total range tested, results of yesterday are valid
- at 420 nm: OD = 0.15 - 3.10
- at 600 nm: OD = 0.01 - 2.26
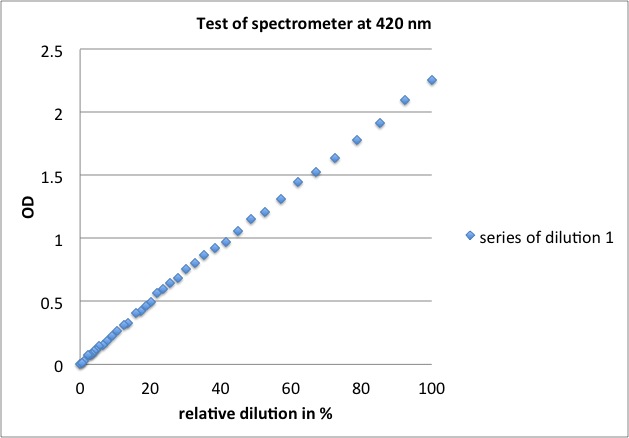
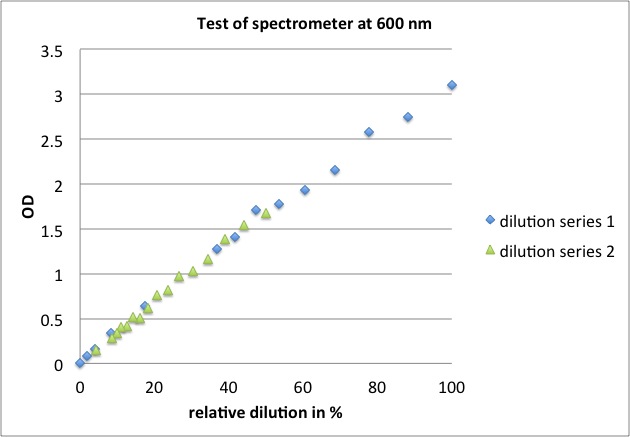
16-09-2011
People: Simon, Anna
Results of 15-09-2011
The plates of the transformation (products of the ligation from 14-09-2011) showed the following amount of colonies:
- Plate 1 (positive control pSB1A3 single cut, old ligase): ca. 150 colonies
- Plate 2 (pSB1C3 + T7promoter, RBS, lacZ, ligation ratio I:V = 1): ca. 200 colonies
- Plate 3 (pSB1C3 + T7promoter, RBS, lacZ, ligation ratio I:V = 0.4): ca. 30 colonies
- Plate 4 (pSB1C3 + blue light sensor, RBS, lacZ, ligation ratio I:V = 1): ca. 1500 colonies
- Plate 5 (pSB1C3 + blue light sensor, RBS, lacZ, ligation ratio I:V = 0.4): ca. 500 colonies
- Plate 6 (vector background pSB1C3): 5 colonies
- Plate 7 (positive control, pSB1A3 single cut, new ligase): ca. 200 colonies
- Plate 8 (insert background blue light sensor, RBS, lacZ): ca. 50 colonies
- Plate 9 (insert background T7promoter, RBS, lacZ): 3 colonies
Cloning
Picking of clones
3 colonies were picked each from plates 2, 3 (Part BBa_K568003 in pSB1C3) and 4, 5 (Part BBa_568002 in pSB1C3) and then inoculated in 5 ml LB Cam each. The cultures were incubated at 37°C over night for miniprep tomorrow.
Cloning of Part BBa_K568004
To ligate the synthetic construct (cut yesterday with E, S) with T7ptag in pSB1C3, the following digestion was done:
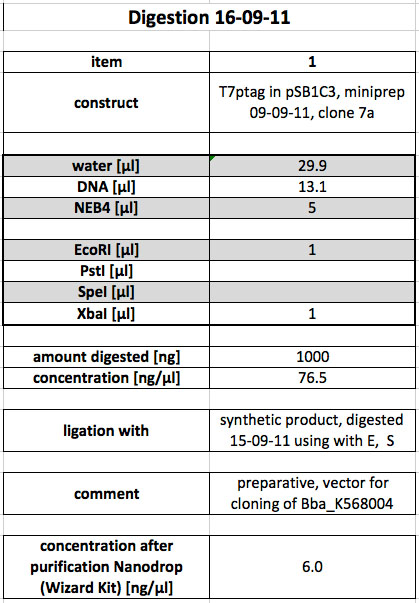
After the digestion was completed, the sample was loaded onto a preparative 1 % agarose gel. After electrophoresis at 120 V, the correct bands were cut out on the transilluminator (365 nm, make sure UV lamp isn't switched on longer than absolutely necessary). Subsequently, the following gel photo was recorded:
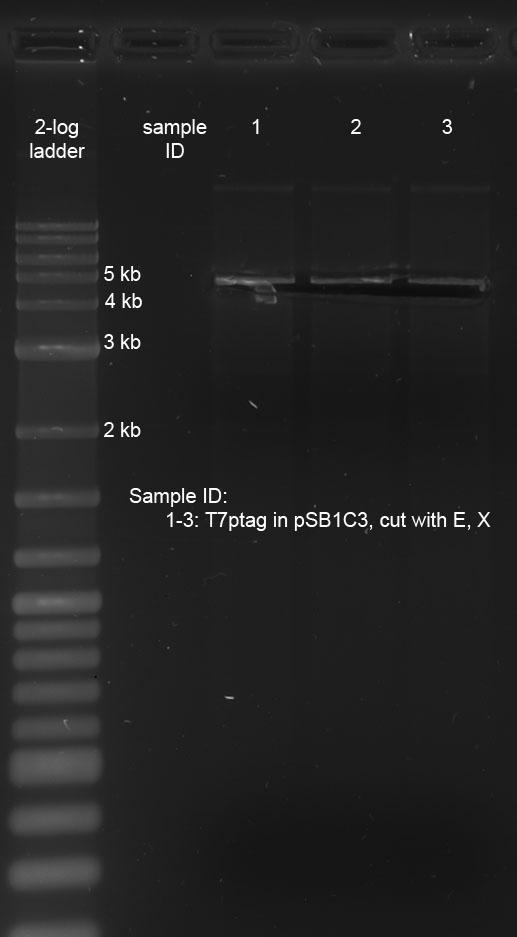
pSB1C3 with T7ptag is 4743 bp long, which corresponds with the location of the cut-out band.
As a next step, the DNA was purified from the gel slice using the Promega Wizard Kit. The concentration of the product was determined to be 6 ng/µl using Nanodrop. This value is not reliable, but for lack of a second, quick way to measure the DNA concentration, the following ligation was perpared using the value nevertheless. Incubation of the Ligation occurred over night at 16 °C.
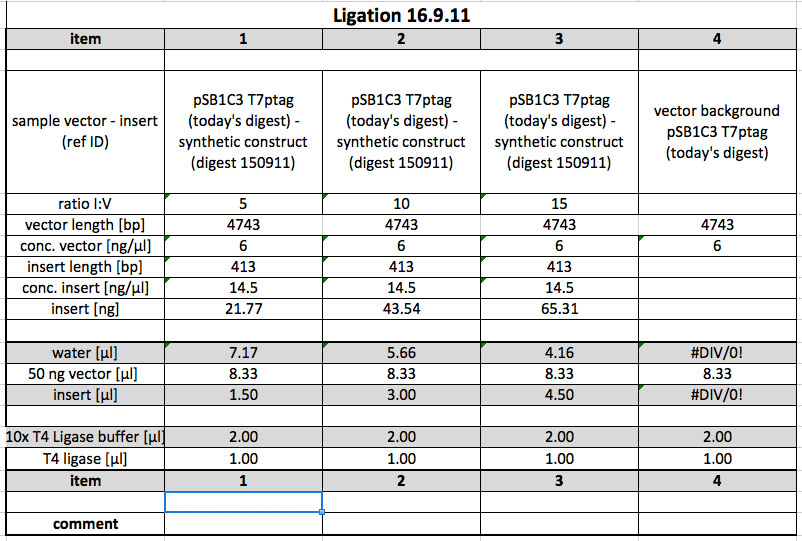
The product of yesterday's ligation was purified using glycogen-ethanol-precipitation (see methods). The product was stored at -20°C for transformation tomorrow.
Other Work
Electrocompetent cells cp919
- new electrocompetent cells (cp919) were made by the following protocol: (it had to be adjusted due to circumstances)
- the day before: 20 ml LB medium was inoculated with 1 µl electrocompetent DH5alpha (from the iGEM registry)
- 450 ml LB medium were inoculated with 4,8 ml of the overnight culture (OD600 = "1,91", goal for start OD600 = 0,02) and incubated at 37°C until a OD(600) of 0,58 was reached
- the medium was distributed on ten falcons and the cells were centrifuged at 4500 rpm for 20 min at 4°C
five falcons broke apart in the centrifuge
- after discarding the supernatant of the five intact falcons, the pellets were resuspended in 15 ml ice-cold ddH2O and distributed on ten tubes.
- the resuspension was centrifuged at 4000 rpm at 4°C for 10 minutes
- after discarding the supernatant the cells were resuspended again in 1 ml of ice-cold ddH2O per tube
- the resuspension was centrifuged at 4000 rpm at 4°C for 20 minutes
- after discarding the supernatant the cells were resuspended again in 1 ml of ice-cold ddH2O per tube
- the resuspension was centrifuged at 4500 rpm at 4°C for 11 minutes
- after discarding the supernatant the cells were resuspended again in 1 ml of ice-cold 10% glycerol per tube
- the resuspension was centrifuged at 4500 rpm at 4°C for 10 minutes
- after discarding the supernatant the cells were resuspended again in 1 ml of ice-cold 10% glycerol per tube
- the resuspension was centrifuged at 4500 rpm at 4°C for 10 minutes
- after discarding the supernatant the cells were resuspended again in 1 ml of ice-cold 10% glycerol per tube
- the resuspension was centrifuged at 4500 rpm at 4°C for 10 minutes
- the cells were resuspended and pooled in a total of 700 µl ice-cold 10% glycerol
- the cells were dispensed in 40 µl aliquots and stored at -80°C for further use (18 tubes)
New media and plates
LB, LB for Agar plates (Cam and Amp) and Luria medium were prepared. Plates were prepared.
17-09-2011
People: Simon, Anna
Cloning
Minipreps of parts BBa_K568003 and BBa_K568002
Yesterday's overnight cultures were prepped today according to protocol. A total of 12 cultures was prepped, 6 of which contained pSB1C3 with K568003 while the other 6 contained pSB1C3 with K568002. After the miniprep, the following concentrations were measured using Nanodrop:
Part K568003 (T7prom, RBS, lacZ):
- 2a: 86.5 ng/µl
- 2b: 71.0 ng/µl
- 2c: 63.5 ng/µl
- 3a: 66.5 ng/µl
- 3b: 88.5 ng/µl
- 3c: 85.0 ng/µl
Part K568002 (blue light promoter, RBS, LacZ):
- 4a: 82.0 ng/µl
- 4b: 78.0 ng/µl
- 4c: 80.0 ng/µl
- 5a: 74.5 ng/µl
- 5b: 97.0 ng/µl
- 5c: 100 ng/µl
Restriction digest and control gel
To check whether the prepped DNA samples contained the correct parts, the following digestion was performed:
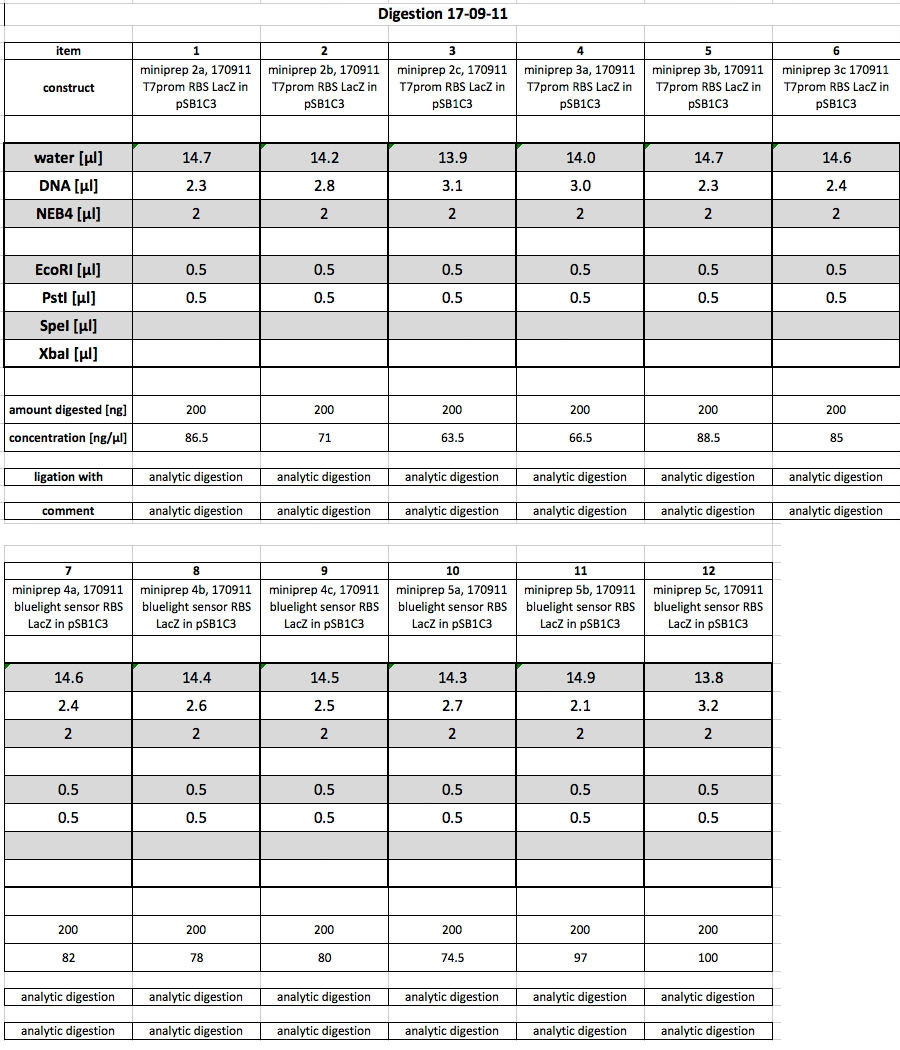
After the digest was completed, the samples were loaded onto a 0.8% agarose gel. After electrophoresis, the following gel photo was recorded:
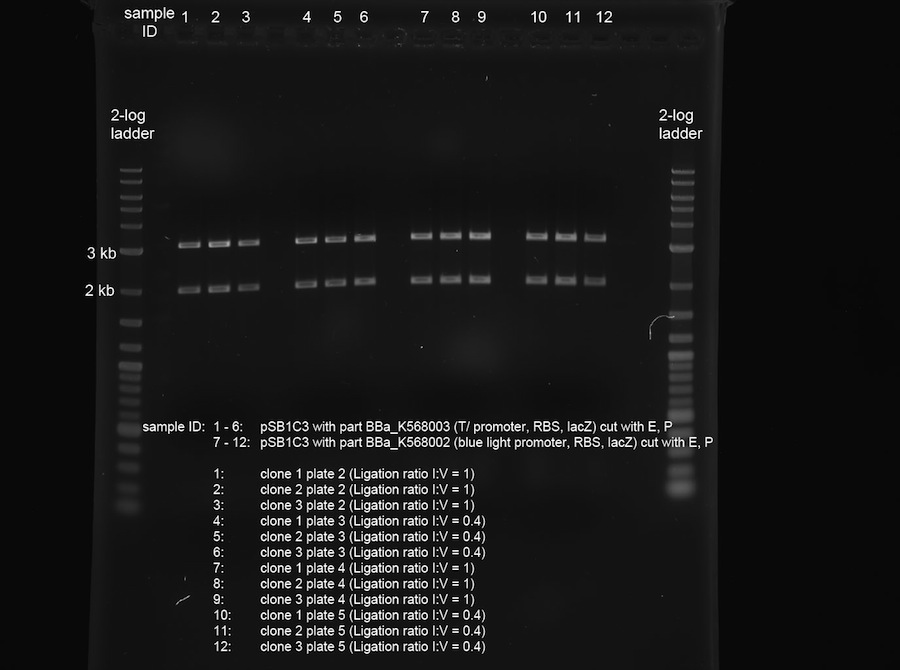
All bands look fine. Samples 1-6 correspond to the minipreps of K568003. Here the insert is 3147 bp and the vector is 2070 bp long. The bands verify this. Samples 7 - 12 correspond to the minipreps of K568002, which has an insert of 3187 bp and the same 2070 bp vector. Again, the bands show that this is correct.
Glycogen/ethanol precipitation
Yesterday's ligation was purified using glycogen/ethanol precipitation. The yielded DNA samples were used for transformation
Transformations
The transformations listed below were carried out. After electroporation the cells were incubated in 1 ml SOC medium for 1 h at 37 °C before plating.
For Redlight Sensor Characterization
All plasmids were electroporated into the new electrocompetent CP919 cells for characterization of the red light sensor later this week.
- Plate Number 1: Part K322127 in pSB1C3 (tube label "K322127 miniprep 04.09.2011"). Plated onto LB Cam plate.
- Plate Number 2: Part K568000 (tube label "5.9.11, 2 vom 1.9, redlight in pSB1C3, 83 ng/µl"). This is our PCR product (redlight sensor). Plated onto LB Cam plate.
- Plate Number 3: cph8 in pSB1A3 (tube label "cph8 Miniprep 4.9.11"). Plated onto both LB Amp.
For generation of Part BBa_K568004
The purified products of yesterday's ligation were electroporated into DH5alpha and plated onto LB Cam plates.
- Plate Number 4: Ligation 1 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 5.
- Plate Number 5: Ligation 2 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 10.
- Plate Number 6: Ligation 3 (16-09-2011), pSB1C3 T7ptag with synthetic construct, ligation ratio I:V = 15.
- Plate Number 7: Ligation 4 (16-09-2011), vector background pSB1C3 T7ptag.
The purified products of the ligation performed on 15-09-2011 were electroporated into DH5alpha, just to make sure that the part is really contained in pSB1A3 and not pSB1C3. Therefore, the transformed cells were plate onto LB Amp and LB Cam plates. The plate Number refers to the LB Amp plate as well as the LB Cam plate in all cases.
- Plate Number 8: Ligation 1 (15-09-2011), positive control pSB1A3 single cut
- Plate Number 9: Ligation 2 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 5.
- Plate Number 10: Ligation 3 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 10.
- Plate Number 11: Ligation 4 (15-09-2011) pSB1A3 T7ptag (purify 1.9.) with synthetic construct, ligation ratio I:V = 15.
- Plate Number 12: Ligation 5 (15-09-2011) vector background pSB1A3 T7ptag (purify 1.9.)
For cloning of I746907 into pSB6A1
We want to use I746907 as a reporter part for our optogenetical AND-gate in e. coli CP919. Therefore the part must be transformed into a low copy vector, since the AND-gate is in pSB1C3 which is a high copy vector. Because CP919 already has a Kan-Resistance, the low copy vector used for this needs to carry Amp-Resistance. That's why we chose pSB6A1, a low copy vector with Amp-Resistance. The DNA was gained from 2011 Kit Plate 1 well 1K by resuspension in 10 µl ddH2O. This was then used for electroporation into DH5alpha which were plated onto LB Amp plates after transformation.
- Plate Number 13: pSB6A1, 0.5 µl DNA used for electroporation
- Plate Number 14: pSB6A1, 1.0 µl DNA used for electroporation
- Plate Number 15: pSB6A1, 8.5 µl DNA used for electroporation
For Testing of I746907
To be able to characterize part I746907, we used a chemical transformation into BL21 DE3 (see methods). 1.5 µl of the DNA were used (c = 63 ng/µl). The transformed cells were plated onto a LB Amp plate.
- Plate Number 16: part I746907 in pSB1A2
Results
sequencing results
(Susan) compared to our parts designed in registry and pSB1C3:
pSB1C3_opt_and-gate_110913_clone_2a_VR-GATC-VR-547731: no results, bad sequencing quality.
pSB1C3_opt_and-gate_110913_clone_6b_VR-GATC-VR-547731: part ok! End of part and plasmid backbone were sequenced.
pSB1C3_synth_construct_110910_clone_5a_VF2-GATC-VF2-545457: Part ok! inserted in backbone
pSB1C3_synth_construct_110910_clone_6b_VF2-GATC-VF2-545457: Part ok! inserted in backbone, 8 bases could not be detected, so part 5a is better, here the whole sequence is right. After looking at the raw data, sequence seems to be ok.
18-09-2011
People: Simon, Flo
Results
The transformations done yesterday worked quite well.
- Plate 1 (Cam): lawn with a few single colonies
- Plate 2 (Cam): lawn with a few single colonies
- Plate 3 (Amp): ca. 20 colonies
- Plate 4 (Cam): ca. 300 colonies
- Plate 5 (Cam): ca. 40 colonies
- Plate 6 (Cam): ca. 20 colonies
- Plate 7 (Cam): 4 colonies
No colonies grew on plates 8 through 12 with Cam. This means that the part was really cloned into pSB1A3. Hence, we use colonies from plates 4 - 6 for miniprepping of part BBa_K568004 in pSB1C3.
- Plate 8 (Amp): > 1000 colonies
- Plate 9 (Amp): no colonies
- Plate 10 (Amp): ca. 900 colonies
- Plate 11 (Amp): ca. 900 colonies
- Plate 12 (Amp): ca. 30 colonies
- Plate 13 (Amp): ca. 50 colonies
- Plate 14 (Amp): ca. 80 colonies
- Plate 15 (Amp): ca. 120 colonies
- Plate 16 (Amp): ca. 300 colonies
Cloning
Picking of clones of Bba_K568004 in pSB1C3 for miniprep
3 clones each were picked from plates 4, 5 and 6 (transformation from 17-09-2011) and 5 ml LB + Cam were inoculated and vortexed shortly for equal distribution of bacterial cells.
Falcon tubes were set almost horizontally in the 37°C-incubator to maximize oxygen input and shaking occurred @ 250 rpm for 3 h and 180 rpm for another 4.5 h.
Miniprep of 3 x 3 clones of Bba_K568004 in pSB1C3
Miniprep done according to kit instruction (Metabion "mi-plasmid mini prep") [See Methods]
Changes:
- All 5 ml bacterial suspension were used for the prep
- Autoclaved water for elution was pre-warmed to ~60°C
Names (according to plate # from 17-09-2011):
- K568004_4-1, c = 64.0 ng/µl
- K568004_4-2, c = 92.0 ng/µl
- K568004_4-3, c = 62.0 ng/µl
- K568004_5-1, c = 67.0 ng/µl
- K568004_5-2, c = 76.5 ng/µl
- K568004_5-3, c = 74.5 ng/µl
- K568004_6-1, c = 75.5 ng/µl
- K568004_6-2, c = 61.5 ng/µl
- K568004_6-3, c = 74.5 ng/µl
Other work
Picking of clones
A clone of plate 16 (BL21 DE3 with I746907) was picked and inoculated in 5 ml LB Amp for testing tomorrow.
2 clones were picked each from plates 12 - 15 (DH5alpha containing pSB6A1) and inoculated in 5 ml Amp for miniprep tomorrow.
Preparation of iGEM-parts for shipping

19-09-2011
People: Simon, Anna
Our Biobricks will be picked up by FedEx today between 1 pm and 3 pm for shipping to the registry. 6 out of 7 Biobricks are ready for take-off, but BBa_K568004 has to be finished in time!
Cloning
Part BBa_K568004
The 9 prepped samples of BBa_K568004 have to be checked for correct sizes of vector and insert. The following digestion was prepared:
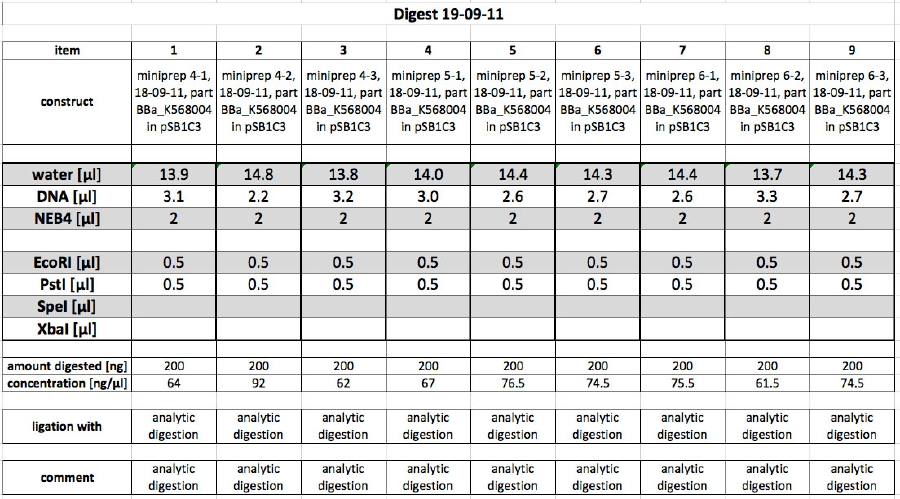
Due to time reasons, 10 µl of the sample was digested for 1 min in the microwave at 800 W, while the other 10 µl of the sample were incubated at 37 °C.
The product of quick digest in the microwave was separated using electrophoresis with an analytical 0.8 % agarose gel. This yielded the following picture:
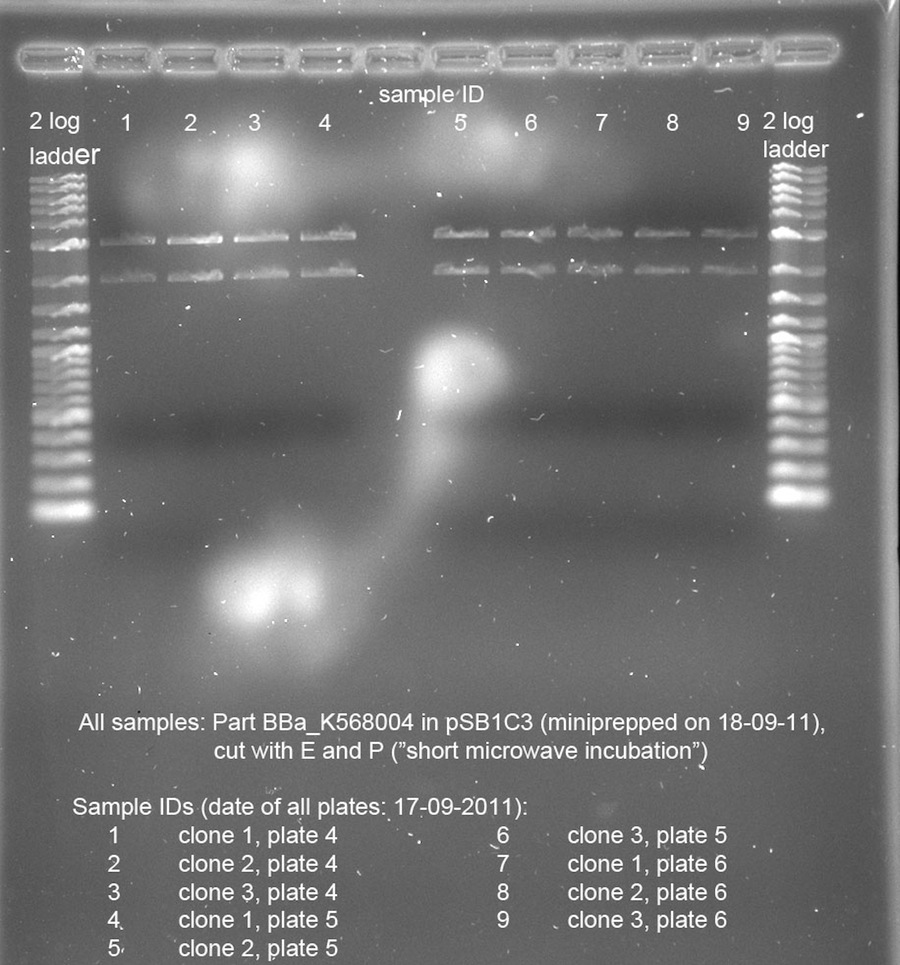
The bands of all samples look fine. The insert BBa_K568004 is 3069 bp long, the pSB1C3 vector is 2070 bp long. All samples show these bands. We chose sample 8 for shipping, because it looked "cleanest" on the gel.
The product of the "normally" incubated restriction digest (1 h at 37 °C) was also loaded onto an analytical 0.8 % agarose gel. The subsequent electrophoresis resulted in the following picture:
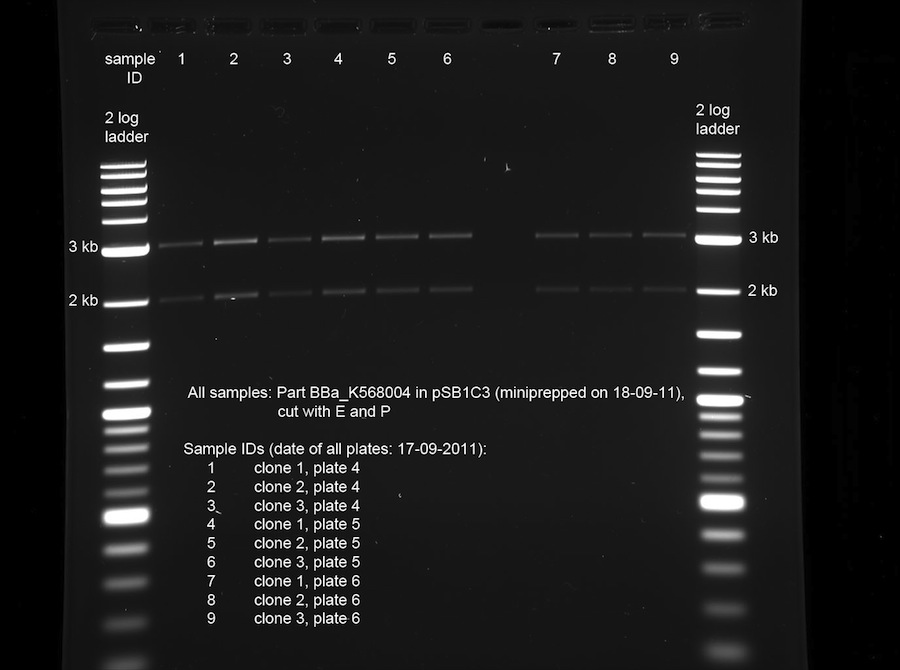
Again, the bands are at the expected positions.
Miniprep of pSB6A1
All six overnight cultures of pSB6A1 in DH5alpha were miniprepped according to metabion's protocol. The cell pellets were pink, indicating that the cells contained the desired plasmid, which carries an RFP-coding sequence as a standard insert. Due to a mistake, the pellet of one sample was lost.
The concentrations were measured using Nanodrop:
- Sample 1: c = 93.0 ng/µl
- Sample 2: c = 79.0 ng/µl
- Sample 3: c = 77.5 ng/µl
- Sample 4: c = 86.5 ng/µl
- Sample 5: c = 102 ng/µl
Other Work
We prepared the miniprepped pSB1C3 with K568004 for shipping. For used amount see the table found in labbook on 18-09-2011.
Preparations for testing of red light sensor tomorrow:
1 clone was picked from each of the following plates (all plates from transformations performed on 17-09-2011):
- Plate 1 (K322127), inoculated in 5 ml LB Cam
- Plate 2 (K568000), inoculated in 5 ml LB Cam
- Plate 3 (cph8), inoculated in 5 ml LB Amp
Testing
GFP Assay
200 µl of the overnight culture of BL21 (DE3) with I746907 (T7 promotor with GFP) in pSB1A2 were inoculated into 5 ml of fresh LB Amp and incubated for 1.5 hours at 37 °C. Before the samples for measurement in the plate reader were prepared, we made a change of medium to M63, because LB shows slight fluorescence itself.
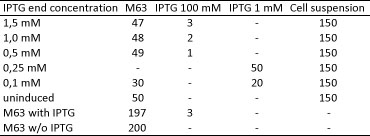
The measurement using the plate reader occured over night, with heating the plate to 37 °C and shaking it between measurements.
Used filters: excitation filter: 485 nm, emission filter: 520 nm
Problem: Fluorescence was measured every 10 minutes while measurement of Absorbance occured only every 100 minutes. This led to spikes in the diagram.
Results
no concentration dependency of GFP induction (between 1.5 and 0.1 mM IPTG), as IPTG concentration was in saturation range
this table shows measured values with
- calculated: Fluorescence/Absorbance
- mean of triplicate measurement
- adjusted to 100 %
- corrected for outliers
- shortened to 700 min
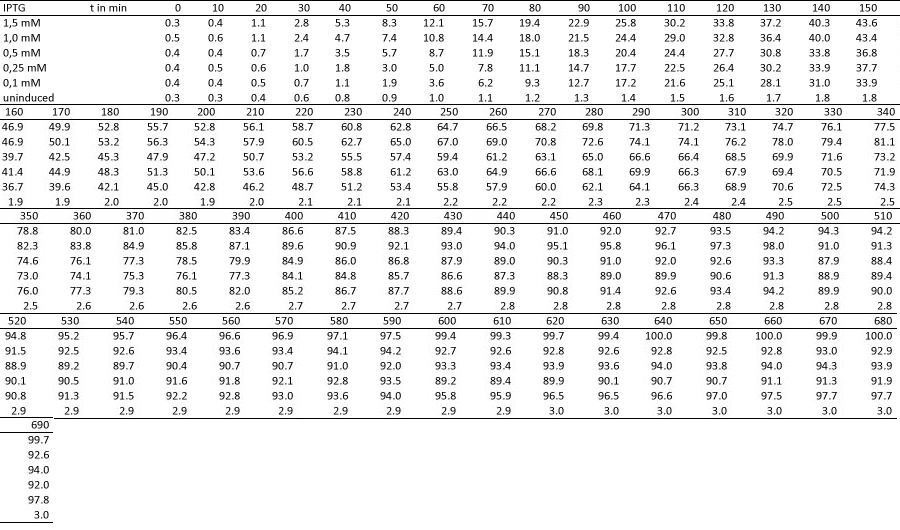
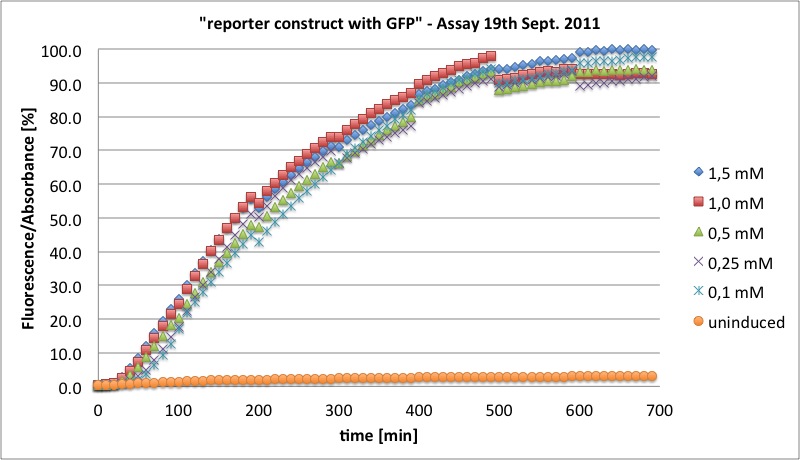
ideas for new GFP Assay:
- much lower IPTG concentrations
- measurement of Absorbance every ten minutes
20-09-2011
People: Simon, Anna
Cloning
To be able to test the optogenetical AND-Gate in CP919, we need to transform the desired reporter part into a low copy vector. Therefore, BBa_ I746907 will be ligated into pSB6A1.
Digest
The following digest was performed:

After this, the samples were separated by electrophoresis using a preparative 1 % agarose gel. The correct gel bands were cut out on the transilluminator at 365 nm (keep irradiation time as short as possible). After this, the following photo was recorded:

The bands match the expectations.
Samples - : The cut out band around 4 kb is the cut vector pSB6A1, which is 4022 bp long. The band visible at around 1 kb is the RFP coding sequence (1069 bp).
Samples - : Part I746907 is 924 bp long. This corresponds to the cut out bands. The vector pSB1A2 can be seen at 2 kb (it is 2079 bp long).
Ligation
After purification of the DNA from the gel slices using the Promega Wizard Kit, the following ligation was prepared and incubated at 16 °C over night:

Testing
Miller Assay for testing red light sensors
Comparision of the parts:
- our red light sensor K568000, the PCR product of K322127, pSB1C3 (C) in cp919
- K322127 of the team Edinburgh 2010, pSB1C3 (C) in cp919
- cpH8 in pSB1A3 in cp919 as a negative control
three lighting options:
- off light (710 nm)
- on light (626 nm)
- dark
light sensitive cultures were grown in the dark for five hours prior to the experiment
OD600 around 2.5 (of cultures before dilution at the start of the experiment), dilution in LB medium to about OD = 0.1
Start of Assay: - measurement of Abs(600nm)
- new eppi with 0,5 ml of Zbuffer + 20 μl of freshly prepared 0,1% SDS+ 40 μl of Chloroform (under fume hood) + 420 µl LB Medium + 20 µl sample
- 0,1 ml of the samples are taken and transferred to the Zbuffer
- mix the solution by vortexing for 10 s (all samples equal vortexing time)
- cenrrifugation: 1 min, RT, 4000 rcf
- transferring of 100 μl supernatant to 96 well plate (for photometer)
- initiation of assay with 20 μl of ONPG (4mg/ml) NOTE START TIME
- incubation at 37 °C
- stop reaction with 50 μl of 1 M Na2CO3 NOTE STOP TIME
- measurement of Abs (420nm) and Abs (550nm)
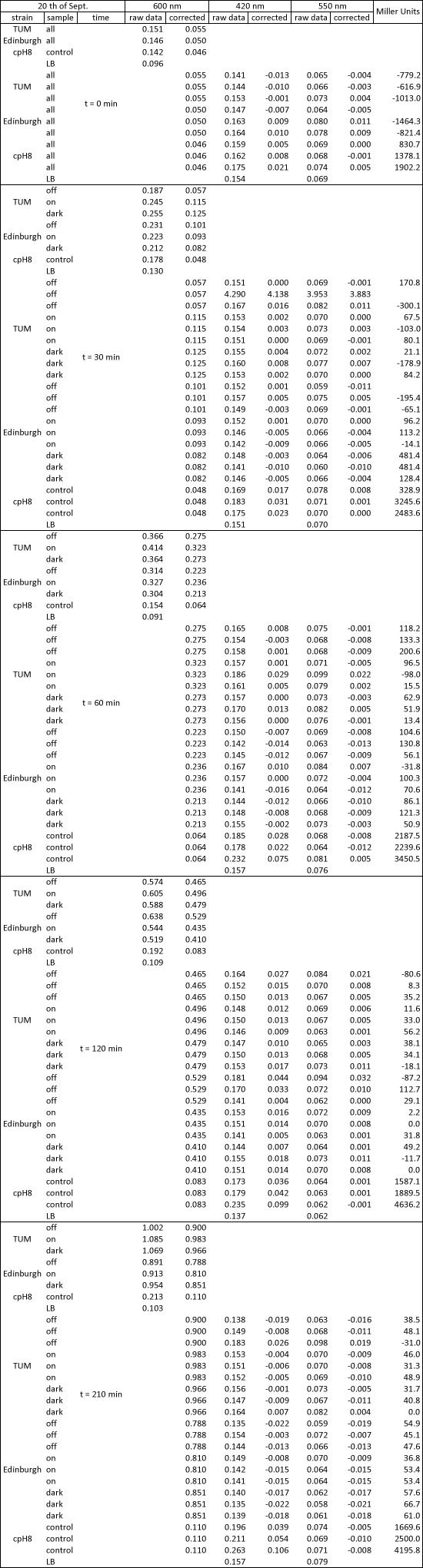
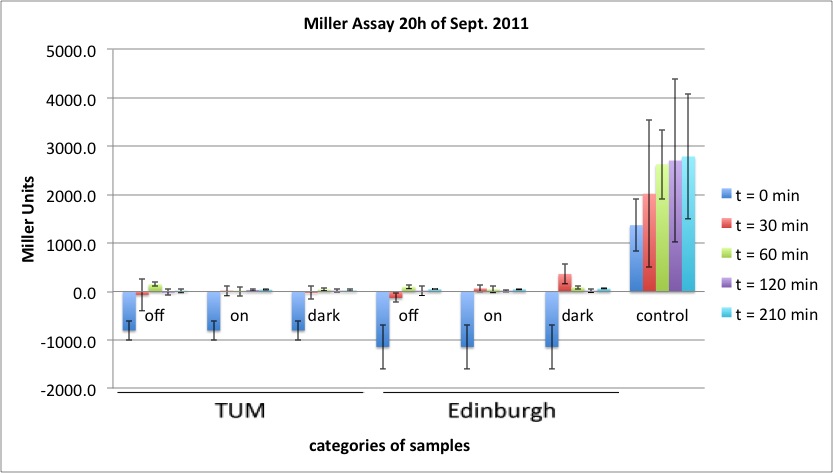
Results
In none of the cells with either the K568000 or the K322127 lacZ could be detected.
possible improvements for further experiments:
- make sure, the cells are in log phase, diluting down earlier, e.g. at a OD600 of about 0.7, however cells shouldn't be in lag phase after 210 min, so at least the last values represent lacZ expression in log phase
21-09-2011
Wiki freeze day!
Yesterday's ligation was incubated at 65 °C for 10 minutes to heat inactivate the enzymes. The samples were stored at -20 °C and can be used for transformation, as soon as we get to characterizing our optogenetical AND-Gate (BBa_568001). We still hope to generate some data to characterize this part in the near future...