 "
"
Team:Glasgow/Results/Nissle
From 2011.igem.org
| Line 5: | Line 5: | ||
<h2>Aims</h2> | <h2>Aims</h2> | ||
<p> | <p> | ||
| + | -To find a transformable, biofilm forming chassis | ||
| + | </br> | ||
-To prove that <i>E.coli</i> Nissle form biofilms | -To prove that <i>E.coli</i> Nissle form biofilms | ||
</br> | </br> | ||
| Line 18: | Line 20: | ||
</br> | </br> | ||
| - | <h2></h2> | + | <h2>Methods</h2> |
| + | <p>Since our project was to examine the dispersal or fixation of a biofilm, we were in need of a chassis that had the ability to form them. Originally <i>Pseudomonas aeruginosa</i> was considered as a chassis, but it was quickly discarded. We found our chassis in <i>E.coli</i> Nissle 1917. It is non-pathogenic, and unlike <i>P.aeruginosa</i> it does not require a shuttle vector. However, the cells were not readily available, and had to be extracted from <i>Mutaflor</i> Tablets. We selected for the desired cells using MacConkey's agar </p> | ||
| + | <p>To prove biofilm formation glass slides were put into 50ml tubes containing 20ml of LB broth. The LB covered around a third of the glass slide, this is the area where the biofilm would form. The LB was then inoculated with 20μl of fresh overnight culture of <i>E. coli</i> Nissle. These tubes were then left on a bench top shaker at room temperature for 24 and 48 hours. The glass slides were then removed and the biofilm was stained by standard gram stain method. Photographs were taked of the stained biofilms.</p> | ||
| + | <p>Biofilm formation was also confirmed by SEM pictures that showed the extracellular matrix, such as Picture 1 below.</p> | ||
| + | <div> | ||
| + | <img src="http://farm7.static.flickr.com/6177/6166139079_a35d6a5930_m.jpg"/><p><font size="1" color="grey">Picture 1: SEM image of Nissle biofilm showing the extracellular matrix</font></p> | ||
| + | <p>Once it had been shown that <i>E.coli</i> Nissle formed biofilms, a time series of biofilm formation was performed. The tubes containing the slides were left on the tabletop shaker for a set amount of time (time points ranged from 1hr to 24hrs). After the biofilm had grown its alloted time the glass slide was carefully removed and placed into a fresh 50ml tube with 25ml of LB (which completely covered the biofilm) and left to allow the bacteria to disperse for 1hr. The slide was then transferred to a fresh 50ml tube with 25ml of LB. The biofilm is scraped off using a spatula, and the resulting culture was vortexed for 1 minute to break up any residual chunks of biofilm. The culture containing the dispersed cells and the culture containing the broken-up biofilm were then plated in serial dilution to obtain an estimate of the number of cells in each culture. The results are shown below.</p> | ||
| + | <div><img src="http://farm7.static.flickr.com/6164/6168708841_f4b9970e7c_z.jpg" alt="Graph of percentage dispersal from biofilm"> | ||
| + | <p><font size="1" font color="grey"> Graph 1: The total percentage of cells dispersed when a biofilm is transferred into new media vs time in hours </font></p> </div> | ||
| + | |||
| + | <p> The data also showed that over time the amount of cells firmly within a biofilm increases at a similar rate to those that disperse and are not attached to the biofilm.</p> | ||
| + | |||
| + | <div> | ||
| + | <img src="http://farm7.static.flickr.com/6171/6169259636_24e606eb08_z.jpg" alt="Cell count of biofilm and planktonic cells vs time"> | ||
| + | <p><font size="1" color="grey"> Graph 2: The estimated number of dispersed cells compared to the number of biofilm cells vs time in hours</font> | ||
| + | </p> | ||
| + | <p>Chemically and Electrically competent <i>E. coli</i> Nissle cells were made and successfully transformed with RFP. Electroporation was notably more efficient than any chemical transformation. Problems were caused by the cells' tendency to clump together, which we assume is caused by their long fimbriae that were visible on the SEM pictures. | ||
| + | this problem can be countered by either vortexing or sonicating the culture before plating them. We expect sonication to be the more efficient method. | ||
</html> | </html> | ||
Revision as of 16:57, 21 September 2011


Results
Aims
-To find a transformable, biofilm forming chassis -To prove that E.coli Nissle form biofilms -To prove that E.coli Nissle are transformable with standard methods -To prove that E.coli Nissle are transformable with standard E.coli biobricks and can express them -To make E.coli Nissle available as a chassis to the Registry of Standard Biological Parts should the above be achieved -To transform our novel biobricks into E.coli Nissle and express them -To operate our entire synthetic system within E.coli Nissle biofilm
Methods
Since our project was to examine the dispersal or fixation of a biofilm, we were in need of a chassis that had the ability to form them. Originally Pseudomonas aeruginosa was considered as a chassis, but it was quickly discarded. We found our chassis in E.coli Nissle 1917. It is non-pathogenic, and unlike P.aeruginosa it does not require a shuttle vector. However, the cells were not readily available, and had to be extracted from Mutaflor Tablets. We selected for the desired cells using MacConkey's agar
To prove biofilm formation glass slides were put into 50ml tubes containing 20ml of LB broth. The LB covered around a third of the glass slide, this is the area where the biofilm would form. The LB was then inoculated with 20μl of fresh overnight culture of E. coli Nissle. These tubes were then left on a bench top shaker at room temperature for 24 and 48 hours. The glass slides were then removed and the biofilm was stained by standard gram stain method. Photographs were taked of the stained biofilms.
Biofilm formation was also confirmed by SEM pictures that showed the extracellular matrix, such as Picture 1 below.

Picture 1: SEM image of Nissle biofilm showing the extracellular matrix
Once it had been shown that E.coli Nissle formed biofilms, a time series of biofilm formation was performed. The tubes containing the slides were left on the tabletop shaker for a set amount of time (time points ranged from 1hr to 24hrs). After the biofilm had grown its alloted time the glass slide was carefully removed and placed into a fresh 50ml tube with 25ml of LB (which completely covered the biofilm) and left to allow the bacteria to disperse for 1hr. The slide was then transferred to a fresh 50ml tube with 25ml of LB. The biofilm is scraped off using a spatula, and the resulting culture was vortexed for 1 minute to break up any residual chunks of biofilm. The culture containing the dispersed cells and the culture containing the broken-up biofilm were then plated in serial dilution to obtain an estimate of the number of cells in each culture. The results are shown below.
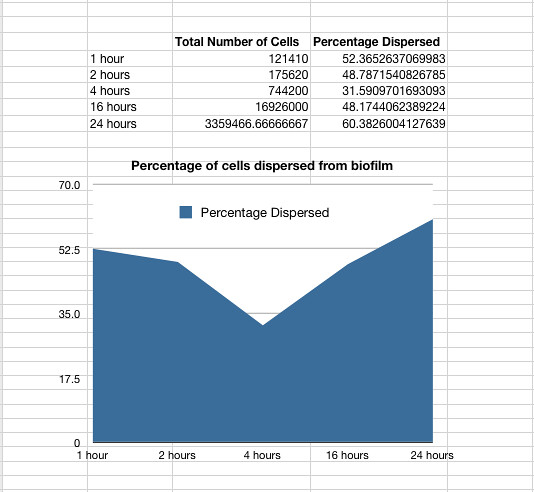
Graph 1: The total percentage of cells dispersed when a biofilm is transferred into new media vs time in hours
The data also showed that over time the amount of cells firmly within a biofilm increases at a similar rate to those that disperse and are not attached to the biofilm.
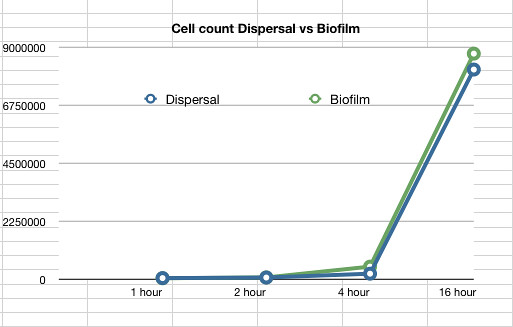
Graph 2: The estimated number of dispersed cells compared to the number of biofilm cells vs time in hours
Chemically and Electrically competent E. coli Nissle cells were made and successfully transformed with RFP. Electroporation was notably more efficient than any chemical transformation. Problems were caused by the cells' tendency to clump together, which we assume is caused by their long fimbriae that were visible on the SEM pictures. this problem can be countered by either vortexing or sonicating the culture before plating them. We expect sonication to be the more efficient method.