 "
"
Team:WashU/Notebook/June2011
From 2011.igem.org

June 1
- Lesson from Bert Berla on how to design primers
- Codon Optimized the four genes using tool at: http://www.encorbio.com/protocols/Codon.htm
- Double checked gene and restriction sites to make sure enzyme does not cut in the middle of the gene
- Design Primers for cassettes
- Ura3, Leu2, KanMX4, and NatMX4
June 2
- Made two liters of LB solution
- 25g of LB Broth per Liter (10g Tryptone, 5g Yeast Extract, and 10g NaCl)
- Autoclaved solutions: 30 minute sterilization.
- Refrigerated after cooling.
June 3
- Made YPD
- In 800ml water in 1 L bottle dissolve 10g of BactoYeast extract
- Dissolve 20g of BactoPeptone in the above solution
- In 200ml water in 500ml bottle dissolve 20g Dextrose
- Autoclave both, combine after autoclaving. (30 minute sterilization)
- Made 1000X Ampicillin Stock
- Added 2g Ampicillin to 20ml water.
- Added NaOH until dissolved.
- Sterile filtered.
- Frozen in -20C Freezer
- Made cultures of E. coli strains with KanMx4 and Natmx4 inserts.
- In culture tubes, added 5ml LB and 5ul 1000X Ampicillin Stock.
- Transferred one colony per strain from plates to culture tubes.
- Incubate at 30 degrees Celsius shaking at 225rpm overnight.
- Made cultures of Yeast strains BC177 and BC178
- Added 5ml YPD to two culture tubes.
- Transferred one colony per strain from plates to culture tubes.
- Incubate at 30 degrees Celsius shaking at 225rpm overnight.
June 4
- Making freezer stocks of yeast cultures BC177 and BC178
- Add 1.25mL glycerol to yeast culture
- Freeze yeast culture in -80 degree freezer
- Make E.coli mini-preps for plasmids containing KanMX4 and NatMX4 (Sigma-Aldrich Mini-Prep Kit)
- Transferred 4 ml of each culture to microcentrifuge tubes.
- Centrifuge for one minute at 12000xg
- Pour out supernatant
- add 200uL of resuspension solution with RNase
- add 200uL of lysis buffer
- add 350uL of neutralization buffer
- Centrifuge for 10 minutes at 12000xg
- Insert a Miniprep Binding Column into microcentrifuge tubes and add 500uL of Column Preparation Solution to each miniprep column.
- Centrifuge at 12000xg for 1 minute. Discard the flow-through
- Take the lysis of the cultures resulting from the 10 minute centrifuge and pipet it into the binding column
- Centrifuge at 12000xg for 1 minute. Discard the flow-through
- Add 500uL of Wash Solution 1 to the column and centrifuge at 12000xg for one minute. Discard the flow-through.
- Repeat the previous step
- Add 750uL of Wash Solution 2 (with ethanol) to the column and centrifuge at 12000xg for one minute. Discard the flow-through.
- Centrifuge at 12000xg for one minute.
- Transfer the column to a fresh collection tube and add 100uL of Elution Solution to the column
- Centrifuge at 12000xg for one minute.
- Store the eluate at -20 degrees C
- This procedure was done separately for the plasmids that contain KanMX4 and NatMX4.
June 5
Read absorbances for all four solutions that contain plasmids with our cassettes: Ura3, Leu3, KanMX4, and NatMX4.
Data was obtained using a nano-drop spectrophotometer. Absorbance was observed between 250nm and 280nm.
DNA Concentrations were as follows:
- Ura3: Very high absorbance reading, must be diluted 1/10 in order to get accurate concentration reading.
- Leu2: Very high absorbance reading, must be diluted 1/10 in order to get accurate concentration reading.
- KanMX4: 30.5 ng/uL
- NatMX4: Very low absorbance reading, eluent was discarded.
- Last year's NatMX4 solution: Very high absorbance reading, must be diluted 1/10 in order to get accurate concentration reading.
- Made new culture of NatMX4
- 5ml of TB + 5uL of 1000x Ampicillin
- Incubated overnight at 31 degrees Celsius.
June 6
- LEU2 group
- Make dilution for nanodrop
- Make solution of 5uL LEU2, 45uL DI water
- Concentration: 42.0 ng/uL
- Make dilution for nanodrop
- URA3 group
- Make dilution for nanodrop
- Make solution of 5uL URA3, 45uL DI water
- Concentration: 35.9 ng/uL
- Make dilution for nanodrop
- KanMX4 group
- 30.5 ng/uL (from previous day)
- NatMX4 group
- Performed mini-prep on previous day's culture
- 200uL eluent with concentration of 28.7 ng/uL
- Made a 1/10 dilution of last year's NatMX4 stock. (DNA Concentration: 23.3 ng/ul)
June 7
- Ordered PCR materials and electrophoresis materials from Sigma-Aldrich.
- Designed primers to add homology to left side of cassette and an AvrII site to right side of cassette.
- Primer designs are:
Leu2
- Forward: TTT CCT AGG CCA AAC TGG AAC AAC ACT CAA CCC
- Reverse: ATA TTT AAT TAT TGT ACA TGG ACA TAT CAT ACG TAA TGC TCA ACC TAA TTT CGT GTC GTT TCT ATT ATG AAT TTC ATT TAT
Ura3
- Forward: TTT CCT AGG ACC ACA GCT TTT CAA TTC AAT TCA TCA TTT
- Reverse: AGT ATC ATA CTG TTC GTA TAC ATA CTT ACT GAC ATT CAT AAC CGC ATA GGG TAA TAA CTG ATA TAA TTA AAT TG
KanMX4
- Forward: TTT CCT AGG AGC TTG CCT CGT CCC CGC C
- Reverse:ATG AAC ATA TTC CAT TTT GTA ATT TCG TGT CGT TTC TAT TAT GAA TTT TCG ACA CTG GAT GGC GGC GTT
NatMX4
- Forward: TTT CCT AGG AGC TTG CCT CGT CCC CGC C
- Reverse: GGG TAA TAA CTG ATA TAA TTA AAT TGA AGC TCT AAT TTG TGA GTT TAG TCG ACA CTG GAT GGC GGC GTT
June 8
- Split into four technique groups
- PCR group
- Practice PCR
- Mastermix:
- 10x buffer:10uL
- betaine: 25 uL
- dNTP: 2.0uL
- Primer 1: 2.5 uL
- Primer 2: 2.5 uL
- Taq polymerase: 5 uL
- dH20: 28.0 uL
- Sample 1:
- 3uL MgCl2
- 1uL DNA template
- 16 uL master mix
- Sample 2:
- 4uL MgCl2
- 1uL DNA template
- 15 uL master mix
- Sample 3:
- 5uL MgCl2
- 1uL DNA template
- 14 uL master mix
- Sample 4 (Negative Control):
- 4uL MgCl2
- 15 uL master mix
- Mastermix:
- Practice PCR
94 degrees: 4min
34 Cycles:
- 94 degrees: 30 sec
- 58 degrees: 30 sec
- 72 degrees: 2 min (2kb product)
72 degrees: 5min
4 degrees: hold
Gel Team:
- We made a gel to use tomorrow:
- 1. Added 500 mL 1xTAE and 5g Agarose
- 2. Microwaved until dissolved completely
- 3. Allowed to cool until able to handle and added 9 mL ethidium bromide
- 4. Poured into gel mold and allowed to cool overnight
June 9
Gel Team:
- We ran the DNA the PCR team amplified yesterday
- filled the electrophoresis chamber with 1xTAE
- Added 2 mL loading buffer to each sample
- put ladder and 4 samples into gel
- ran electrophoresis for ~1h
- We imaged the gel
- used UV imaging machine to take picture below



- The desired band was not visible. We determined the problem was we were testing the wrong template DNA
- We made a new batch of gels using 400mL of water and 3.5g of agarose
PCR team:
- We repeated PCR with new template DNA and freshly ordered supplies
Recipe per sample for running plasmid DNA:
- 2.5uL 10x buffer
- 1.25 uL dNTP
- 1.5 uL forward primer 44
- 1.5 uL reverse primer 43
- 16.5 uL dH20
- 0.25 uL Accutaq LA DNA polyermerase
Mastermix:
- 12.5uL 10x buffer
- 6.25 uL dNTP
- 7.5 uL forward primer 44
- 7.5 uL reverse primer 43
- 82.5 uL dH20
- 1.25 uL LA polyermerase
Sample 1, 2, 3:
- 1.5 uL DNA of plasmid pTCP2031v
- 23.5 uL Mastermix
Sample 4:
- 23.5 uL plasmid Mastermix
Recipe per sample for running genomic DNA:
- 2.5uL 10x buffer
- 1.25 uL dNTP
- 0.5 uL DMSO
- 0.5 uL forward primer 42
- 0.5 uL reverse primer 43
- 17.0 uL dH20
- 0.25 uL Accutaq LA DNA polyermerase
Mastermix:
- 12.5uL 10x buffer
- 6.25 uL dNTP
- 2.5 uL DMSO
- 2.5 uL forward primer 42
- 2.5 uL reverse primer 43
- 85.0 uL dH20
- 1.25 uL Accutaq LA DNA polyermerase
Sample 5, 6, 7:
- 1.5 uL vector DNA
- 23.5 uL Mastermix
Sample 8: 23.5 uL genomic Mastermix
Run same temperatures as before with 34 cycles
June 10
- Ran PCR products on a 1% agarose gel.
- Used UV imaging machine to produce picture below
-
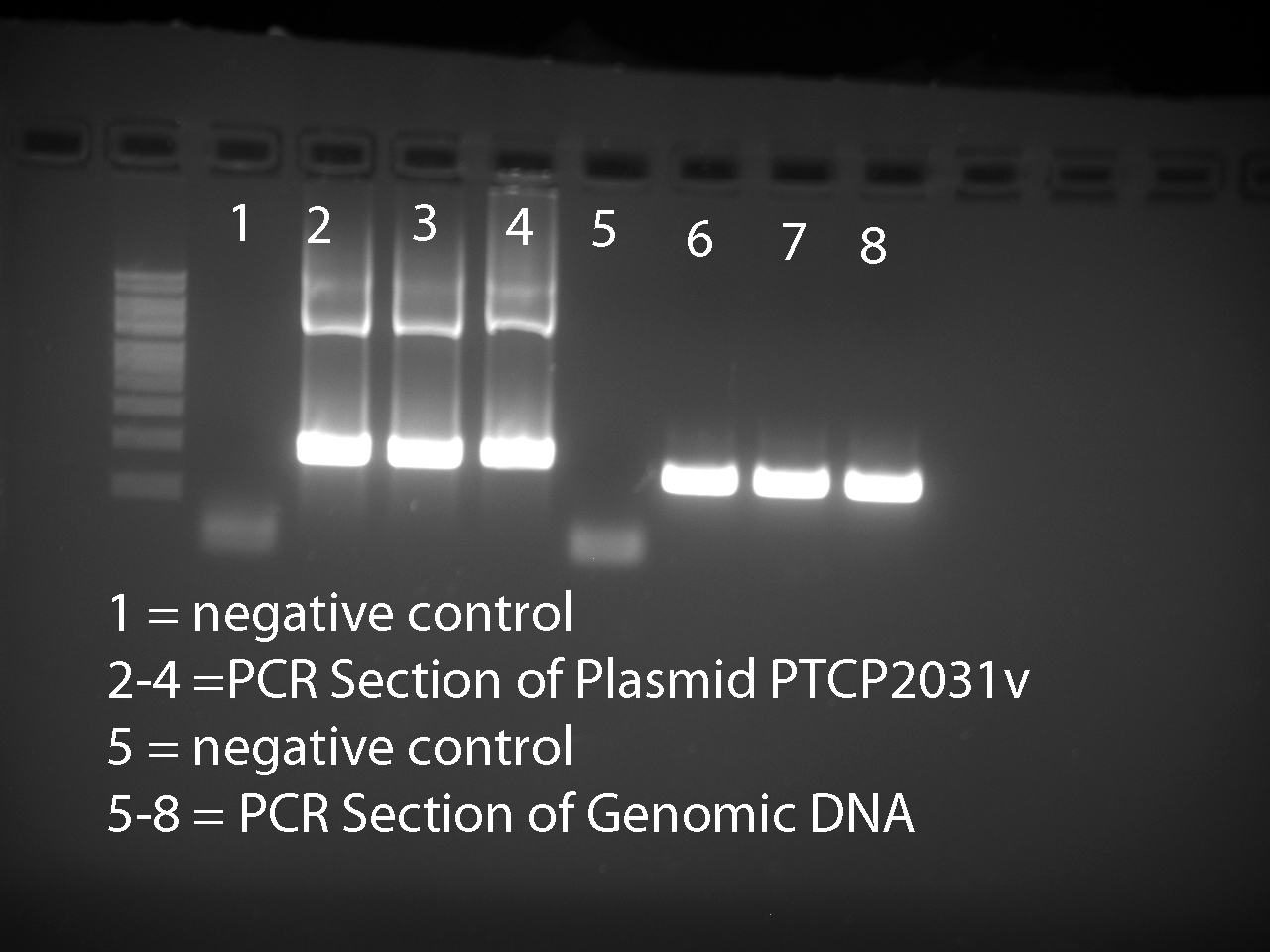

All six expected bands were visible.
Performed gel extraction on all six bands using the Sigma Kit
- excised the bands from the gel. The genomic band was too big so we cut it in half
- vector:.37g
- genomic1: .20g
- genomic2: .21g
- Placed gels in separate collection tubes and added 3 gel volumes of Gel Solubilization Soln. Allowed gels to dissolve
- Prepared 3 binding columns by adding 500uL of Column prep soln and centrifuging for 1 min. Discarded eluate
- Added 1 gel volume isopropanol to each collection tube and mixed thoroughly.
- Loaded the gel solution into the binding columns in 700 uL portions. centrifuged for 1 min and discarded eluate
- Added 700uL of Wash Soln to each column, centrifuged 1 min, and discarded eluate
- Centrifuged columns again for 1 min and discarded eluate
- Transfer each binding column to a fresh collection tube. Added 50uL of Elution Solution and incubate 1 min
- centrifuge 1 min. The elution contains the DNA
June 13
- Performed PCR product clean-up using a Sigma Kit.
gel team:
- we did PCR cleanup
- Added .5 mL of the column prep soln to 2 miniprep column and centrifuged at 12000g for 1 min. Discard eluate.
- Added 65 uL of Binding soln and 15 uL of each PCR reaction to separate columns and centrifuged for 1 min. Discard eluate
- Add .5ml of Wash soln to each column and centrifuge 1 min. Discard eluate.
- Centrifuge additional 2 min. Discard eluate and place binding column into new collection tube
- Add 50uL elution soln and allow to incubate 1 min
- centrifuge 1 min. The PCR product is in the eluate
Nano-dropped four samples:
- Gel extraction vector DNA
- No DNA present; one large peak at 230nm signifying the presence of protein.
- Gel extraction genomic DNA
- No DNA present; one large peak at 230nm signifying the presence of protein.
- PCR product clean-up vector DNA
- 50uL of solution at 36.9ng/uL
- PCR product clean-up genomic DNA
- 50uL of solution at 18.4ng/ul
June 14
Transformation:
- Made plates with LB agar
- Added ~300 mL H2O to graduated cylinder
- Added 12.5 g LB broth, 7.5 g Bacto Agar, and aforementioned water to Erlenmeyer flask
- Stirred on hot plate to boiling
- Added water until total volume was 500 mL
- Autoclaved
- Returned to stir plate for 15 minutes to cool
- Added 0.05 g ampicillin (for antibiotic resistance marker) to flask
- Poured into 20 plates
- 3 plates discarded because the agar had solidified by the time they were poured
Gel:
- Did a restriction digest
- 5 uL 10X Buffer (#2 NEB)
- 30 uL DNA from PCR cleanup of vector DNA
- 15 uL water
- 1 uL 1 ku EcoRI
- incubated at 37 C overnight
June 15
Assay Preparation:
- Make 500ml YPD
- Autoclaved YPD and poured into 16 petri dishes
June 16
Biobrick Transformation in E. coli:
- GC5 E. coli cells and SOC medium arrived
- Began transformation of E. coli with Leu2 and Ura3 biobricks
- Thawed 50 uL cells in ice bath
- Added 10 uL sterile ddH2O into biobrick wells 11A and 11C in plate 3
- Waited 5 minutes for DNA to resuspend
- Transferred the 10 uL to microcentrifuge tubes
- Added 20 uL cells to Ura3 tube, 20 uL cells to Leu2 tube, 10 uL cells to control tube
- Added 2 uL biobrick DNA to respective tubes
- Waited 30 minutes for cells to transform on ice
- Heat shocked in water bath at 37 C for 45 seconds
- Placed cells on ice for 2 minutes
- Added 200 uL SOC medium into each tube (100 uL into control tube)
- Put tubes in shaker at 37 C at 220 rpm for one hour
- Spread onto plates with LB agar (made June 14)
- Using a total of 6 plates, spread 50 uL onto one plate and the rest (172 uL) onto the other, for each of the three tubes
Gel:
- Ran digestion on a gel
- Ran a new digestion
- 30 uL DNA from PCR cleanup of genomic DNA
- incubated at 37 C overnight

Assay Preparation:
- Streak yeast strains, BC177 and BC178, each onto a separate petri dish with YPD
June 17
Tranformation:
- Due to the presence of ampicillin on the plates, we expected no colonies on the control plates, but colonies should grow on the Leu2 and Ura3 plates because they should have antibiotic resistance against ampicillin, if the transformation succeeded. Colonies grew on all plates, even the negative controls; we assume it must have been caused by deficient or faulty amipicillin. Because of this, we cannot determine whether the transformation proceeded correctly or not.
- We are performing a test of our amicillin stock as well as our new ampicillin stock.
- Three culture tubes each with 4 mL of LB. One tube is a positive control (No AMP), one has 4 uL of our old AMP stock, and one has 4 uL of our new AMP stock.
- Cells that do not contain a plasmid are placed in each culture tube.
- Began shaking at 240rpm at 37 degrees Celsius at 10:30 am.
- At 2:30 pm (4 hour incubation) the OD of the positive control was around .250 and both AMP cultures were still around zero.
- CONCLUSION: Ampicillin stocks are viable. Perhaps the reason for the lawns on the negative control transformation plates were due to Ampicillin being added while the media was still too hot.
- We will be making new plates with new ampicillin, following the same procedures as described before:
- Made plates with LB agar
- Added ~300 mL H2O to graduated cylinder
- Added 12.5 g LB broth, 7.5 g Bacto Agar, and aforementioned water to Erlenmeyer flask
- Stirred on hot plate to boiling
- Added water until total volume was 500 mL
- Autoclaved
- Waited for the solution to cool
- Added 500 uL ampicillin to flask
- Poured into 12 plates
- Made plates with LB agar
Gel
- ran practice digest 2 on a gel

PCR Team
- We ran PCR on genomic DNA using two different protocols (Sigma Aldrich's & Bert's) to create more product for the gel team to practice on.
Total samples: 8
Sigma's Protocol:
Recipe per sample for running genomic DNA:
- 2.5uL 10x buffer
- 1.25 uL dNTP
- 0.5 uL DMSO
- 0.5 uL forward primer 42
- 0.5 uL reverse primer 43
- 17.0 uL dH20
- 0.25 uL Accutaq LA DNA polyermerase
Mastermix:
- 12.5uL 10x buffer
- 6.25 uL dNTP
- 2.5 uL DMSO
- 2.5 uL forward primer 42
- 2.5 uL reverse primer 43
- 85.0 uL dH20
- 1.25 uL Accutaq LA DNA polymerase
Sample 1, 2, 3 (not marked):
- 1.5 uL genomic DNA
- 23.5 uL Mastermix
Sample 4(negative sign in circle): 23.5 uL Mastermix
Bert's Protocol (w/adjustments by May):
Recipe per sample for running genomic DNA:
- 2 uL 10x Buffer
- 0.4 uL dNTP
- 0.5 uL forward primer 42
- 0.5 uL reverse primer 43
- 10.35 uL dH2O
- 0.25 uL Accutaq LA DNA polymerase
- 5 uL betaine
Mastermix:
- 10 uL 10x Buffer
- 2 uL dNTP
- 2.5 uL forward primer 42
- 2.5 uL reverse primer 43
- 51.75 uL dH2O
- 1.25 uL Accutaq LA DNA polymerase
- 25 uL betaine
Sample 5,6,7 (marked with a dot):
- 1 uL genomic DNA
- 19 uL Mastermix
Sample 8(negative sign in circle): 19 uL genomic Mastermix
Anneal at 58 degrees with 34 cycles
Assay Preparation:
- Check growth of yeast strains on petri dishes streaked yesterday.
- Concluded colonies on plates were too close together
- Re-streaked two new petri dishes of BC177 and BC178 yeast strains using colonies from yesterday’s petri dishes.
June 18
- Check growth of yeast strains on petri dishes
- Concluded colonies on plates were too close together for plate with strain BC177.
- Re-streaked one new petri dish for BC177
June 20
- Check growth of yeast strains on petri dishes
- Concluded colonies on our new stock plates were isolated enough for streaking
- Isolated one colony from BC177 and BC178 yeast strains and cultured them in test tubes with 5 mL of YPD solution
Tranformation again:
- Thawed 50 uL cells in ice bath
- Added 20 uL cells to Ura3 tube, 20 uL cells to Leu2 tube, 10 uL cells to control tube
- Added 2 uL biobrick DNA to respective tubes
- Waited 30 minutes for cells to transform on ice
- Heat shocked in water bath at 37 C for 45 seconds
- Placed cells on ice for 2 minutes
- Added 200 uL SOC medium into each tube (100 uL into control tube)
- Put tubes in shaker at 37 C at 220 rpm for one hour
- Spread onto plates with LB agar (made June 17)
- Using a total of 6 plates, spread 50 uL onto one plate and the rest (172 uL) onto the other, for each of the three tubes
Gel Team:
- Did PCR cleanup on PCR product from 6/17
- ran a digestion of PCR product from 6/17 with .3 uL EcoRI
- ran a gel of the digestion
- Did gel extraction of PCR product from 6/17

PCR Team:
Ran PCR on the four cassettes (Leu2, Ura3, KanMX4, and NatMX4) with the newly arrived primers. This is amplify the cassettes, add homology to the insertion site, and add a restriction site. Sigma's Protocol from June 17 was used. Leu2 and Ura3 were annealed at 50 degrees C while NatMX4 and KanMx4 were annealed at 55 degrees C.
June 21
Transformation:
- The transformation procedure performed yesterday worked. The Leu2 and the Ura3 plates all had some colonies on them, while the control plates had none.
- Today, we made cultures of the Leu2 and Ura3 colonies.
- Added 5 mL LB and 5 uL ampicillin to two tubes each.
- Using sterile technique, picked off a colony from one plate and added it to one tube. Did same for other.
- Incubated overnight on shaker at 30 C.
PCR: We used the Sigma protocol with an annealing temperature of 58 degrees C on the sample plasmid DNA provided to us by Burt. This was in order for the gel team to practice ligation after cutting with an restriction site.
Gel:
- ran gels of PCR product from 6/20
- made new gels
- 350 mL TAE and 3 g agarose
- ran gels of PCR product from 6/21
- [[1]]
_copy.jpg)
_copy.jpg)
June 22
Transformation:
- Today, we used a plasmid miniprep kit to extract the plasmids from the cultured cells
- For each tube, we extracted 4000 uL (4 mL) cell solution and added 1000 uL each into an individual microcentrifuge tube (for a total of 8 tubes)
- Centrifuged at 12,000 g for 1 minute
- Discarded supernatant
- Added 50 uL resuspension solution containing RNAse A into each of the eight tubes
- Vortexed to resuspend
- Combined the eight tubes into two new microcentrifuge tubes (for Leu2 and Ura3)
- Added 200 uL lysis buffer into both tubes and inverted to mix
- Added 350 uL neutralization/binding buffer and inverted to mix
- Centrifuged at 12,000 g for 10 minutes
- Inserted a binding column into a microcentrifuge tube and added 500 uL to the column
- Centrifuged new tubes 12,000 g for 1 minute; discarded flow-through
- Transferred supernatant (lysate) into columns
- Centrifuged 1 minute and discarded flow-through
- Added 500 uL wash solution to column; centrifuged again and discarded flow-through
- Added 750 uL second wash solution to column; centrifuged again and discarded flow-through
- Centrifuged again and discarded flow-through to remove ethanol
- Transferred to new collection tube
- Added 100 uL elution solution and centrifuged again
- We measured the DNA concentration of the resulting eluate using a Nanodrop.
- Leu2: 329.8 ng/uL
- Ura3: 84.1 ng/uL
PCR:
We repeated the PCR done on June 20th on the cassettes. The only difference is we added a sample with just plasmid DNA and no primers in order to determine if the top band on the gel resulting from the June 20th PCR was the plasmid DNA.
June 23
Transformation:
- The Leu2 concentration was very high. To be sure, we measured the concentrations of both again.
- Leu2: 323.9 ng/uL
- 260/280: 1.89
- 260/230: 2.33
- Ura3: 83.4 ng/uL
- 260/280: 1.99
- 260/230: 2.23
- Leu2: 323.9 ng/uL
- We also began practicing transformation of yeast using two strains that Kim gave us and the pRS425 plasmid. To transform the yeast, we would have to extract the plasmid from the E. coli colonies we were given using a miniprep kit. We cultured the three plates.
- For each plate, we chose one colony and added it to a glass tube of solution. For the two yeast strains, we used 5 mL YPD and 5 uL ampicillin. For the E. coli, the tube contained 5 mL LB only.
- The three glass tubes went into a 30 C shaker overnight.
Assay:
- Practiced using Olis spectrophotometer and computer program for future experiments involve spectrophotometrically assaying for the intermediates and products of the beta carotene pathway.
- Repaired stock beta carotene solution and 1/128 stock beta carotene solution, both in acetone.
- Took preliminary spectra of beta carotene solutions, starting from the stock solution, successively diluting by twofold each time.
Gel:ran a gel of the PCR product from 6/22
_copy.jpg)
_copy.jpg)
June 24
Transformation:
- Due to a misunderstanding, our yeast cultures were accidentally dumped, so we could only work with the E. coli with pRS425 plasmid today.
- Using the miniprep procedures previously described, we miniprepped one tube of the plasmid.
- Nanodrop results:
- Concentration: 235.4 ng/uL
- 260/280: 1.88
- 260/230: 2.30
PCR:
Repeated the PCR from June 22 because we realized the concentration of DNA was too high. For this sample 10 ng of DNA were loaded for Leu2, Ura3, Kan, and Nat.
Assay:
- Experimentally obtained the absorption spectrum of yeast solution in YPD and beta carotene dissolved in acetone at varying concentrations. Started with solutions of each and successively diluted solutions twofold until absorption peaks are no longer characteristic of the solution.
Gel:
- Did a restriction digest on kan and nat cassettes
- ran a gel with leu2 and ura3 plasmids transformed from biobricks and restriction digest product
- picked up gel extraction kits from Cohen lab
- Used spin column gel extraction kit to extract restriction digest products from gel
_copy.jpg)
June 27
Transformation:
- Made YPD plates with ampicillin
- 1.65 g Bacto Agar into 200 mL YPD; autoclaved
- 250 uL ampicillin
- Poured into plates (had to throw out a few and ended up with 4 plates)
- We could not perform the transformation procedure today because we were required to leave the lab early.
- Put the cells into 50 mL YPD (from cultures overnights made yesterday)
- Added 2 mL cell solution into 5 mL more YPD and put in the 30 C shaker overnight
Gel:
- ran a ligation reaction overnight of the restriction digest products from 6/24
June 28
Transformation:
- We cannot use the plates made yesterday because ampicillin is not a marker for yeast; threw them out and planned to plate the yeast with Leu2, the real yeast marker for the plasmid pRS425
- Yeast transformation today
- Transferred 60 uL cells to 50 mL YPD
- Left the cells in the 30 C shaker for approximately 6 hours (checked the OD several times, but it was too low each time; the OD had to be between 0.6-1.2)
- Spun the cells at 3000 G for 5 minutes
- Discarded the supernatant
- Added 25 mL sterile water to each of the two tubes (825, 827)
- Spun again at 3000 G for 5 minutes
- Discarded supernatant
- Resuspended in 1 mL 100 uM lithium acetate to break open cells
- Put the cells into two microcentrifuge tubes each (one control and one transformation; four microcentrifuge tubes total)
- Put in the 30 C shaker for 15 minutes
- Found salmon sperm DNA and put it into a thermocycler, heating 25 uL of it to 98 C and the flash-cooling to 4 C
- Centrifuged the cell solutions for 30 seconds at 3000 G
- Pipetted out supernatant
- Added 240 uL of PEG 3350 to each tube
- Added 5 uL of salmon sperm DNA to each tube
- Added 34 uL sterile water to control tubes and 34 uL DNA to transform tubes
- Added 36 uL 1 M lithium acetate to each tube
- Vortexed
- Left on 30 C shaker for 30 minutes
- Left in 42 C water bath for 40 minutes
- Spun tubes at 1000 G for 30 seconds
- Pipetted out supernatant
- Added 800 uL YPD to each tube and resuspended by pipetting
- Left overnight at room temperature with a book on top to prevent tube caps from popping off
Gel:
- ran a gel of PCR product from 6/24
- ran a gel of our ligation from 6/27
- [[2]]
- did a new restriction digest on nat casette using AgeI
- ran a gel of restriction digest
- [[3]]
- Used spin column gel extraction to extract the DNA from our restriction digest gel
_copy.jpg)
_copy.jpg)
June 29
PCR Protocol for Plasmids Review
- 2.5uL 10x buffer
- 1.25 uL dNTP
- 16.5 uL dH20
- 0.25 uL Accutaq LA DNA polyermerase
- 1.5 uL forward primer
- 1.5 uL reverse primer
- 10ng DNA (1uL if using a diluted sample)
PCR on the Nat ligation product:
- Nat was not detected on a gel but may be because a very small amount of product was made.
- Well 1,2,3 are PCR mix with Nat primers and 3uL of Nat ligation product per well
- Well 4 is positive control with Kan primers and Kan DNA
- Well 5 just has 3uL of Nat ligation product and no primers.
June 30
Diluted gene primers
crtYB F: 10uM, 576uL added
crtE F: 10uM, 193uL added
crtI F: 10uM, 1.028mL added
CCD1 F: 10uM, 1.614mL added
Gene R: 100uM, 1.337mL added
There is also a working solution of 10uM of gene R.
Gel:
- Ran a gel of PCR product from 6/28
- Did a restriction digest of nat casette using AgeI
- ran ligation reaction on gel
- used PCR cleanup kit on part of digest product and used in a ligation reaction, ran reaction overnight
.jpg)
_copy.jpg)
![[1]](https://3626779877665202800-a-1802744773732722657-s-sites.googlegroups.com/site/washuigem2011/files/practicegel4.jpg?attachauth=ANoY7cqWrQR283tB0AfD093BqtqdYD1wl1Y5BK1IGGhIGm_cST9h-5_TU2zuCjX9q9MUIBaSFkZmdrtTzGvbQlWYVlbv5dfiVHs2z-wcUi5koAvGLZw9TBtEsxEoKlopsYIE98Mv81nZSpB0WdrjLa7LkG8jLDJ59dMvyD9EQBSKtAtcF3pvGH8BvUMy7GXvIgZew32deAn9MO5iwhLAcHiu2w8NKPGA0Q%3D%3D&attredirects=0){kind=link}
![[2]](https://3626779877665202800-a-1802744773732722657-s-sites.googlegroups.com/site/washuigem2011/files/practiceligation1.jpg?attachauth=ANoY7cqnDhncwPaHrLoyouAca8p_S9mnMvYohAATaeuKRAbQA0UoWLEtQieXFFXDMk9896sNdntrQFUwJLw_OdaVn7w2yhxg3i-UDZZlcXHGonPdkrvABnCikENBu0A6qBl18TIEtR_dHlTJSflI3Ikywth7SSYIv7TNPxG7uFQXhAU_svEZcySCPRK92btGWgpktOX60n3qvsdgjbXxXkpJkRSo3_QvXw%3D%3D&attredirects=0){kind=link}
![[3]](https://3626779877665202800-a-1802744773732722657-s-sites.googlegroups.com/site/washuigem2011/files/28.jpg?attachauth=ANoY7crPjoCsafhMlC2Xc47CuljJr1kkDXioWVrppI-f6y4xYAYxhwd9u0JV5HWTK6L1yLCqQ9mN_jXi1DAfnm3953SVC5UIXoOd_jDs25jAZbtWDHVwzDEpcqvTp4j98h5H-sQMpQFTScABn_qSEVXMBqMgqUiuSgaHTvWotpkAQ7cHh2Kxw7tnv7avQmbcz3GPLkWxQwmY&attredirects=0){kind=link}
![[4]](https://3626779877665202800-a-1802744773732722657-s-sites.googlegroups.com/site/washuigem2011/files/ligation6-30.jpg?attachauth=ANoY7cpBokuR47bmHt2K9iGXBxGOJ0bXQ0DzEkpPb1WwD9daKCMwCJwrH3Lnt_H3dSc39hqYTLvy_-SStLDTKx88Lp06ruInSiwEtaZecYaIwTA1WeAbRaubMPX0a6UT2jYmS6jz9FH-mmnYOVJatuIn0tPSHJC6Puvk1em-ejFSPjsM96jB7VUYfkCJUhaywEOwSf3-sHHSS0hllGT4RQeo7efxXzlPQg%3D%3D&attredirects=0){kind=link}